原位脱羧制备的配合物(HP2W18O62)(C6H 6NO2)5·2H 2O:结构、光谱及电化学性能
2013-09-15朱鹏飞张雅琴郭枫晚祁晓婷
魏 振 朱鹏飞 张雅琴 郭枫晚 祁晓婷 王 娟*,
(1湖北大学化学化工学院,武汉 430062)
(2武汉大学化学与分子科学学院,武汉 430062)
0 Introduction
Polyoxometalates (POMs)are a unique class of ligands characterized by their discrete size,high symmetry,large anionic charge,aqueous stability,and various redox activities[1-4].Due to these features,POMs have been found to be extremely versatile inorganic building blocks for the construction of supramolecular architectures with potential applications in catalysis,medicine, materials, molecular conductivity,geochemistry and nuclear wasteprocessing[5-8].Hydrogen bonding interactions and other weak intermolecular interactions between small molecules are the welldeveloped methodstoincreasestructural dimensionality of supramolecular systems in the crystal engineering“toolbox”[9-10].While some supramolecular frameworks are open-frameworked and there are guest species occluded by the open-framework host in the structure.Water molecules are the commonly encountered guest species and usually play an important role in the stabilization of the host.
Pyridine-2,6-dicarboxylic acid(H2PDA)is a very important carboxylate derivative.A systematic study of complexes based on H2PDA has been undertaken,which gives biological activities and unique coordination configuration for PDA[11-14].However,H2PDA is not stable under higher reaction temperature.Herein,we report the synthesis of a heteropolyacid complex(HP2W18O62)(C6H6NO2)5·2H2O.The crystal structure,spectroscopy and electrochemistry behavior of the obtained compound are studied.
1 Experimental
1.1 Materials and physical measurement
All materials were of analytical grade and were used without further purification.
Deionized water was used throughout.Elemental analysis (C,H,and N)was performed on a Vario Micro Cube element analyzer.The infrared spectrum was recorded using KBr pellet on a Spectrum One Fourier Transform Infra-red Spectrometer in 4 000~400 cm-1.The electronic absorption spectra were taken on a Shimadzu UV-240 spectrophotometer.Cyclic voltammograms were obtained on a model CHI660C electrochemical analyzer (CH Instruments,Austin,TX,USA)controlled by a personal computer at room temperature.A three-electrode system was used for the measurements,with a bare GCE (3 mm diameter)or C-Ni/GCE used as the working electrode,a saturated calomel electrode (SCE)as the reference electrode and a platinum wire as the auxiliary electrode.All experiments were performed at the laboratory temperature.
1.2 Synthesis of the title compound
A mixture of Na2WO4·2H2O(0.989 6 g,3.0 mmol),H3PO4(0.3 mL,85%),pyridine-2,6-dicarboxylic acid(0.066 8 g)and H2O (15 mL)was stirred for half an hour in air,and pH value of the solution was adjusted to 3.8 by addition of NaOH (0.5 mol·L-1)solution.The mixture was then transferred to a Teflon-lined stainless steel autoclave (25 mL)and kept at 170℃for 6 d.After the autoclave had cooled to room temperature,light green crystals were filtered off,washed with distilled water,and air-dried.Anal.Calcd.for C30H35N5O74P2W18:C,7.17%;H,0.70%;N,1.39%.Found:C,6.74%;H,1.60%;N,1.45%.
1.3 Single crystal structure determination
Single-crystal X-ray crystallographic analyses of 1 with approximate dimension of 0.22 mm×0.24 mm×0.28 mmwasperformed at291(2)Kon a Bruker SMART APEX CCD sealed tube diffractometer with graphite monochromated Mo Kα (0.071 073 nm)radiation.Data collection,indexing,and initial cell refinements were carried out using SMARTsoftware[15].Frame integration and final cell refinements were carried out using SAINT software[16].Absorption corrections for each data set were applied using SADABS program[17].Structure analysis shows that the crystal of monoclinic system P21/n space group,crystal cell parameters:a=1.553 3(2)nm,b=2.006 3(3)nm,c=2.759 9(4)nm,β=103.981(2)°,Z=4,F(000)=8 816.All calculations were performed by using SHELXTL-97 program[18].The crystal parameters,data collection,and refinement results are summarized in Table 1.Selected bond lengths and angles are listed in Table 2.More details on the crystallographicstudies as well as atom displacement parameters are presented in the Supporting Information.
CCDC:905785.
2 Results and discussion
2.1 Description of the crystal structure
Singlecrystal X-ray diffraction analysisrevealsthat compound 1 consists of five 2-picolinic acid,one heteropolyanion of[HP2W18O62]5-and two water molecules(Fig.1).In the heteropolyanion,central P atomsare surrounded by oxygen atoms to form two sets of PO4tetrahedron.The P-O distance is 0.152 8~0.162 2 nm,while the O-P-Oangle varies from 103.9°~113.9°,Comparing the O-P-O bond angles with that of regular tetrahedron (113.0°),the PO4in 1 has been distorted greatly.W-O distance is 0.163 7~0.239 6 nm,O-W-O angle varies from 71.16°~174.65°.
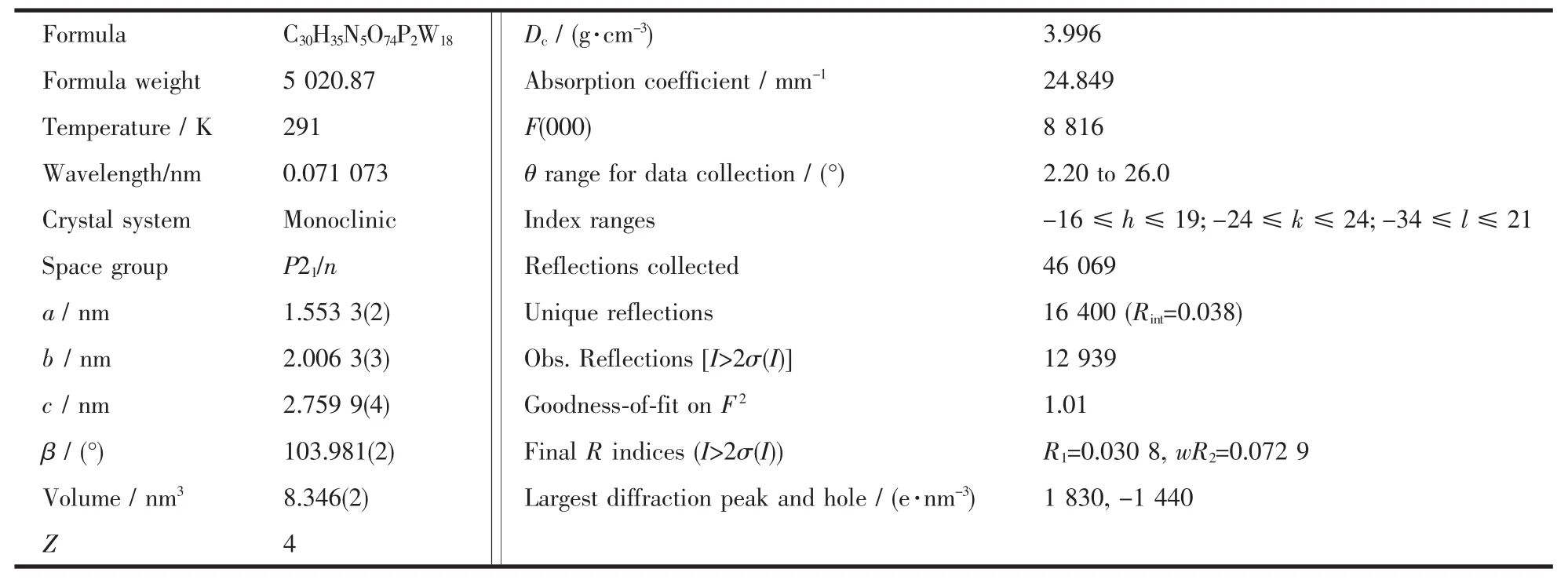
Table 1 Crystal data and structure refinement for the title compound
There are two categories of W atoms:six at polar positions and twelve at equator positions.Oxygen atoms in[HP2W18O62]5-can be divided into four groups according to their coordination number[19]:Ot(terminal oxygen atoms connecting only one W atom),Ob(oxygen atoms located in the share corners between two W3O13units),Oc(oxygen atoms connecting edge sharing WO6octahedrons in the same W3O13unit)and Oa(oxygen atoms connecting the P heteroatom and W atoms).Relevant W-O bond distances in the anion can be classified into three groups:W-Ot0.163 7 ~0.175 1 nm,W-Ob0.178 0~0.204 5 nm,W-Oc0.231 0~0.236 8 nm,W-Oa0.233 3~0.239 6 nm.Their mean bond distances are 0.183 4(6)nm,0.194 8(7)nm,and 0.233 5(6)nm,respectively.While W-O-Wbond angles vary from 89.8(2)°~162.6(4)°.The results indicate that PO4tetrahedron and WO6octahedron has distorted a little attributed to the force interaction between 2-picolinic acid and heteropolyanion.
To our surprise,the pyridine-2,6-dicarboxylic acid (H2PDA)is decarboxylated into 2-picolinic acid in the reaction,which may be due to higher reaction temperature.Besides,the nitrogen atom of 2-picolinic acid is protonated.The average bond length of N-H is 0.085 9 nm.The interesting structural feature of compound 1 is that the interactions between the inorganic and organic moieties are mainly hydrogen bonding.All the protonated 2-picolinic acid are with a total five positive charges balanced the negative charges on polyanions[HP2W18O62]5-to make compound 1 electrically neutral.
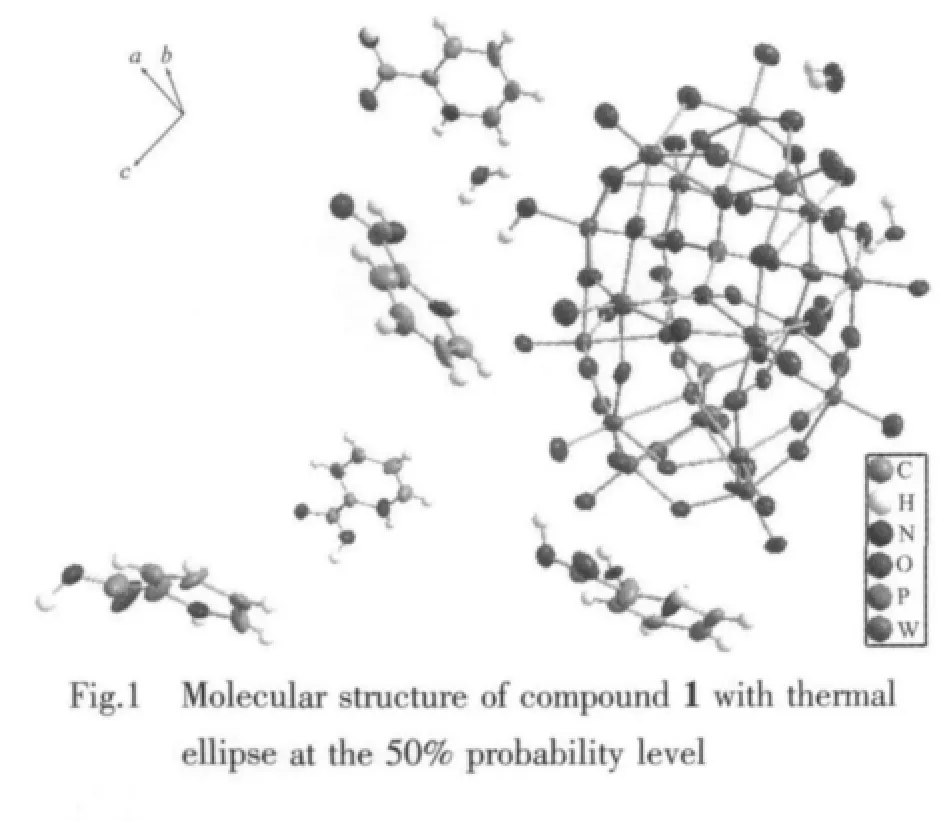
As seen in Fig.2a,along the a axis,the polyoxoanion and 2-picolinic acid are polymerized to form a 1D chain through weak interactions(N2-H2K…O14 0.3063nm)and hydrogen bonding(N2-H2K…O19 0.2848nm).In the same way,the structure is extended into a 2D network down the b axis (O1…H4C-O4W 0.297 0 nm,O52 … H4D-O4W 0.250 8 nm).The network then is stacked into a 3D supramolecular framework via weak interactions of two 2-picolinic acids and two polyanions(see Fig.2(b)).It is interesting that two carboxyl groups of 2-picolinic exist in a “chair” conformation via weak interactions of O63 and O64 (0.297 9 nm,Fig.3).The related hydrogen bond data are listed in Table 3.
2.2 IR spectrum
The IR spectrum of compound 1 exhibits four characteristic peak of Dawson heterropolyanion.The four are at 1 090 cm-1attributed toν(P-O),958 cm-1attributed to ν(W-Od),907 cm-1attributed to ν(W-Ob),and 780 cm-1attributed to ν(W-Oc).Comparing the IR spectrum of compound 1 with its precursor acidα-H6P2W18O62·n H2O,the vibration peak of ν(W-Od)is red shifted from 961 cm-1to 958 cm-1,and theν(W-Ob)shifted from 912 cm-1to 907 cm-1,while the peaks of ν(P-O)and ν(W-Oc)are basically the same.These results indicate that the heterropolyanion in the complex still retains the Wells-Dawson structure.In addition,bands in the 1 227~1 732 cm-1region are ascribed to characteristic vibration of 2-picolinic acid.Furthermore,the IR spectra show a strong and wide band at about 3 460 cm-1,which is most likely ascribed to the stretching vibration of the crystal water molecules.
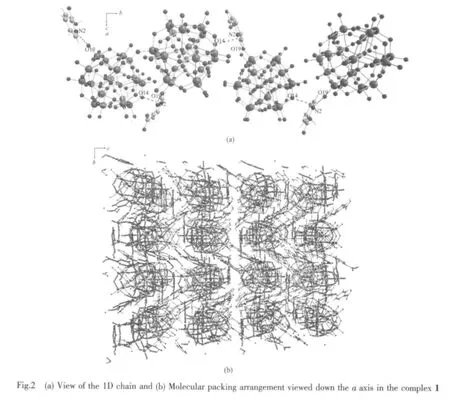
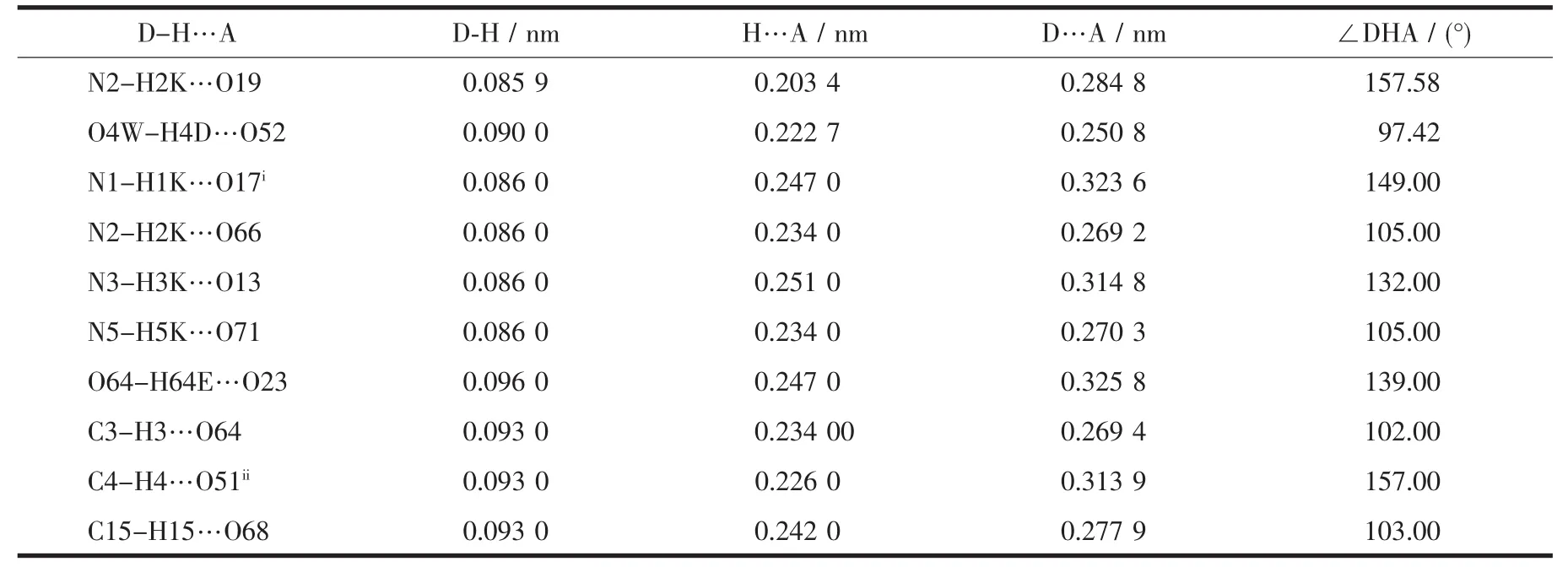
Table 3 Selected hydrogen-bonding geometry for the title compound
2.3 UV spectrum
The characteristic absorption peaks of free Wells-Dawson type polyanion of α-H6P2W18O62·n H2O are at 210 and 325 nm[20].The UV peak for free 2-picolinic acid is at 262 nm[21].The UV spectrum of compound 1 in water exhibits two absorption peaks at 234 nm and 293.8 nm.The former is strong,which can be ascribed to the charge-transition absorption of Ot→W.A wide broad peak around 260~340 nm suggests that a new conjugated system has been created between the organic and inorganic moieties.
2.4 Electrochemical behavior
Cyclic voltammetric measurement of the complex was carried out in deionized water at 25℃in H2SO4(0.1 mol·L-1)at a scan rate of 100 mV·s-1.In the potential range from 1.0 ~1.5 V,four couples of reduction and oxidation were recorded(As shown in Fig.4).
For reversible process(ΔEp<59 mV),according to the formula|Ep-Ep/2|=56.5/n,the three step reaction electron transfer number can be calculated to 2,1,1 respectively.For an irreversible system(ΔEp>59 mV),in the light of formula|Ep-Ep/2|=47.7/(αn)(α=0.5)[22],the last step reaction electron transfer number n can be calculated to 2.In accordance with reference[23],the first wave for single electron peak is diffusioncontrolled,while the third and fourth reductions are accompanied by protonations.However,in our case,two electronic peaks are seen around the first peak position.This may because that H2SO4makes it converted into two electronic peaks.The last irreversible process is two-electronic peak with protonation asreported by others[24].The four reversible redox waves are summarized as shown in equations(1)~(4):
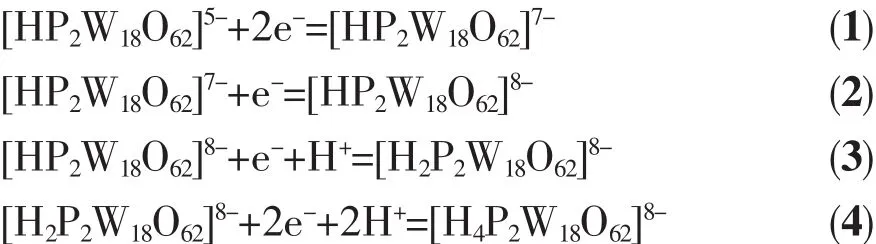
3 Conclusions
The single crystal compound (HP2Wl8O62)(C6H6NO2)5·2H2O (1)was synthesized by using a hydrothermal method.The compound belongs to monoclinic system,P21/n space group.The crystal cell parameters are a=1.553 3(2)nm,b=2.006 3(3)nm,c=2.759 9(4)nm, β=103.981(2)°,Z=4,F(000)=8 816.IR,UV and elemental analysis confirm the crystal structure.The complexes with electrochemical activity can also be used for REDOX catalytic reaction.The gain and loss electronic reactions are as follows:
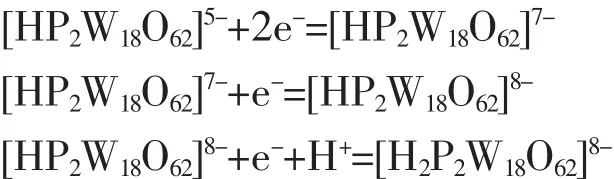
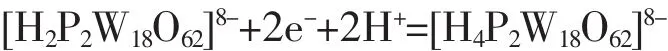
Supplementary materials:Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication No.905785.Copies of the data can be obtained free from the Director,CCDC,12 Union Road,Cambridge CB2 1EZ(Fax:t44 1223 336 033;E-mail:deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
[1]Fan L L,Xiao DR,Wang EB,et al.Crystal Growth&Design,2007,7(4):592-954
[2]Pope M T.Heteropoly and Isopolyoxometalates:Vol.8.Berlin:Springer-Verlag,1983.
[3]Upreti S,Ramanan A.Crystal Growth&Design,2006,6(9):2066-2071
[4]Santiago R,Pablo V,Luis L,et al.Inorg.Chem.,2003,42:3709-3711
[5]Ednéia C,JoséA D,Sílvia CL,et al.Microporous Mesoporous Mater.,2010,132(1/2):103-111
[6]Sun C Y,Liu S X,Liang D D,et al.J.Am.Chem.Soc.,2009,131(5):1883-1888
[7]Vanpelt C E,Crooks W J,Choppin G R.Inorg.Chim.Acta,2003,346:215-222
[8]Wang J P,Wei M L,Niu J Y.Trans.Met.Chem.,2004,29:81-85
[9]Raghuraman K,Katti K K,Barbour L J,et al.J.Am.Chem.Soc.,2003,125(23):6955-6961
[10]Liu M S,Yu Q Y,Cai Y P,et al.Crystal Growth&Design,2008,8(11):4083-4091
[11]Liu J Z,Zhang Z,Yin X Hong,et al.J.Chem.Crystallogr.,2012,42:1001-1006
[12]Zafar A S,Mohd K,Sarvendra K,et al.Eur.J.Med.Chem.,2010,45:264-269
[13]Liu L F.J.Inorg.Organomet Polym.,2012,22:1308-1313
[14]Wang J,He F L,Wang X Y,et al.J.Coord.Chem.,2011,64(13):2312-2320
[15]SMART,Version 5.624;Brüker AXS,Inc.:Madison,WI,2000.
[16]SAINT,Version 6.36A;Brüker AXS,Inc.:Madison,WI,2000.
[17]Sheldrick G.SADABS,Version 2.10,University of Göttingen:Göttingen,Germany,2003.
[18]Sheldrick G.SHELXTL SHELXTL-97:Program for Crystal Structure Refinement,University of Göttingen,Germany,1997.
[19]Niu J Y,Wang Z L,Wang J P.Inorg.Chem.Commun.,2003,6:1272-1274
[20]Zhao J W,Yu L,Wang J P,et al.Chin.J.Appl.Chem.,2005,12:1324-1328
[21]Huang J,Xu Y H,Chen X K,et al.J.Rare Earths,2012,30(6):586-591
[22]Moreira A B,Oliveira H P M,Atvars T D Z,et al.Anal.Chim.Acta,2005,539:257-261
[23]Sadakane M,Steckhan E.Chem.Rev.,1998,98:219-237
[24]WANG Chong-Tai(王崇太),HUA Ying-Jie(华英杰),SUN Zhen-Fan(孙振范),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2011,27(2):473-478
