前列癃闭通片的定性定量方法研究
2013-09-11管燕,肖遐
管 燕 ,肖 遐
(1.湖南中医药大学附属第二医院药剂科,湖南 长沙 410005;2.湖南师范大学医学院药学系,湖南 长沙 410013)
前列癃闭通片由前列癃闭通胶囊剂型改革而来[1],由淫羊霍、黄芪、虎杖、枳壳、桃仁等十一味中药组成。具有益气温阳,活血利水功效。用于肾虚血瘀所致癃闭,症见尿频,排尿延缓、费力,尿后余沥,腰膝酸软;前列腺增生见上述证候者。因胶囊制备用的提取物为浸膏,胶囊剂在使用、存放过程中容易吸湿。不利于药效的发挥。故将其改为片剂后,采用薄膜包衣工艺,使产品的防潮性能大大提高,能有效地保障产品的疗效,并可增加临床用药的选择性,方中黄芪具有补气,利尿,抗病原微生物的作用[2];淫羊藿对病原微生物有杀灭或抑制作用,有促进尿液分泌的作用[3],为了更好的控制本片剂的质量,本文采用薄层色谱法对方中黄芪、虎杖、枳壳进行定性鉴别,用HPLC 法对淫羊霍中有效成分淫羊霍苷进行含量测定,为该制剂建立质量标准提供依据。
1 仪器与试药
仪器:岛津LC-10ATvp 高效液相色谱仪;HW2-000 色谱工作站。日本SHIMADZU CIV-240IPC 紫外分光光度计;BO211D 电子分析天平(西德Sartorious)。
试药:前列癃闭通片(自制),各阴性对照品(自制),黄芪甲苷标准品、虎杖对照药材柚皮苷标准品、淫羊藿苷标准品均为中国药品生物制品检定所提供;试剂:乙腈均为色谱试剂,水为重蒸水,其它试剂均为分析纯。
2 薄层鉴别
2.1 黄芪的薄层鉴别试验
取本品10 片,除去包衣后,研成细粉,加甲醇40mL,加热回流2 小时,滤过,滤渣用10mL 甲醇洗涤,洗液并入滤液中,水浴浓缩至约10mL,置已处理好的中性氧化铝柱中,用40%甲醇50mL 洗脱,收集洗脱液,蒸干,残渣加水10mL 使溶解,加水饱和的正丁醇振摇提取3 次,每次20mL,合并正丁醇液,加氨试液振摇提取2 次,每次20mL,弃去氨试液,正丁醇液蒸干,残渣加甲醇1mL 使溶解,作为供试品溶液[4]。另取黄芪甲苷对照品,加甲醇制成每1mL 含1mg的溶液,作为对照品溶液。再取除黄芪外的其他处方量药材,按制备工艺制成缺黄芪的阴性样品,同法制成阴性样品溶液。照薄层色谱法(中国药典2010 版一部附录ⅥB)试验[5],吸取上述三种溶液各10μL,分别点于同一硅胶G 板上,以三氯甲烷-甲醇-水10℃以下放置分层的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃烘至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;缺黄芪的阴性样品无干扰,结果见图1。
2.2 虎杖的薄层鉴别试验
取本品10 片,除去包衣后,研成细粉,加乙醇30mL,浸渍2 小时,滤过,滤液蒸干。残渣加乙醇1mL 使溶解,作为供试品溶液。另取虎杖对照药材1g,加乙醇15mL,同法制成对照药材溶液。照薄层色谱法(中国药典2010 版一部附录ⅥB)试验,吸取上述溶液各5μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G 薄层板上,以正已烷-醋酸乙酯(4∶1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同的橙黄色荧光斑点,阴性对照无干扰,结果见图2。
2.3 枳壳的薄层鉴别试验
取本品5 片,除去包衣后,研成细粉,加水5mL湿润,加正丁醇30mL 超声提取40 分钟,滤过,滤液用水饱和的正丁醇振摇提取2 次,每次20mL,合并正丁醇液,蒸干,残渣加甲醇1mL 使溶解,作为供试品溶液。另取,加甲醇制成每1mL 含0.5mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2010 版一部附录ⅥB)试验,吸取上述溶液各5μL,分别点于同一硅胶G 薄层板上,以醋酸乙酯-甲醇-水-二乙胺(10∶1 .7∶1.3∶0.3)为展开剂,展开,取出,晾干,喷以三氯化铝试液,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,阴性对照无干扰,结果见图3。
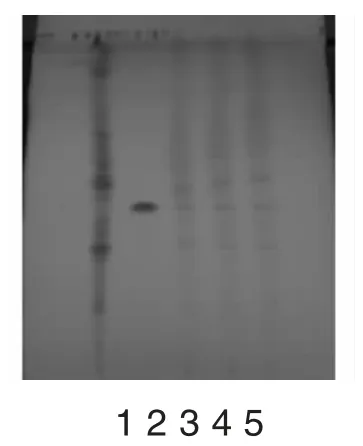
图1 黄芪的薄层色谱
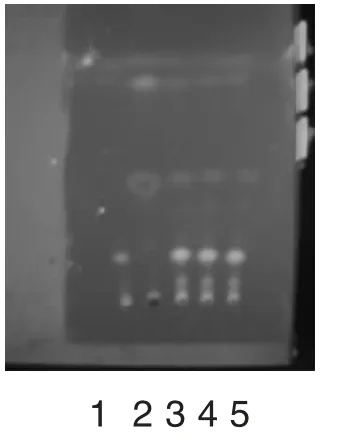
图2 虎杖的薄层色谱
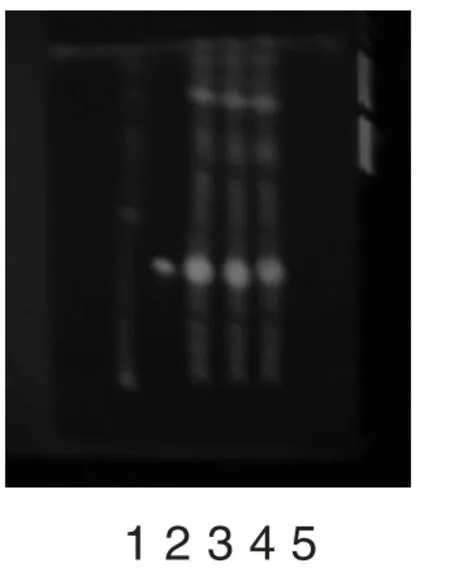
图3 枳壳的薄层色谱
3 含量测定
本品中含淫羊藿,其功效为补肾阳、强筋骨、祛风湿,其主要有效成分是淫羊藿苷,故参照中国药典2010年版一部淫羊藿项下淫羊藿苷的含量测定方法,对本品中淫羊藿苷的含量进行测定,试验结果表明,缺淫羊藿的阴性溶液无干扰,且样品分离效果好。经方法学考察,本含量测定方法结果较好,方法可行。经测定十批成品,其平均含量为0.45mg/片,故暂规定本品每片中含淫羊藿苷不得少于0.36 mg。
3.1 色谱条件
色谱柱:C18(Microsorb-MV 100-5 250mm×4.6mm,5μm)
流动相:乙腈-0.1%磷酸溶液(26:74)
检测波长:270nm
流速:1.0mL/min。
在选定条件下,淫羊藿苷峰与样品中其它组分色谱峰可达基线分离,且与其它相邻色谱峰分离度大于1.5;按淫羊藿苷峰计算,理论板数在2000以上。
3.2 供试品溶液的制备
参照中国药典及相关文献,本品用稀乙醇为提取溶媒进行超声提取,现对超声提取时间进行考察,方法如下。
取本品研成细粉,取三份各约0.4g,精密称定,置具塞三角瓶中,分别精密加入稀乙醇20ml,称定重量,第一份超声处理(120W,40KHz)15 分钟、第二份超声处理30 分钟、第三份超声处理45 分钟,放冷,称定重量,加稀乙醇补足减失的重量,摇匀,滤过,取续滤液10μl,注入液相色谱仪中,测定,结果见表1。

表1 提取时间考察结果
结果表明:超声处理15 分钟时,样品中的淫羊藿苷提取不完全,超声处理30 分钟与45 分钟时,其含量相当,均可将样品中的淫羊藿苷提取完全,因此确定超声处理时间为30 分钟。
供试品溶液制备方法:取本品10 片,精密称定,研细,取细粉0.4g,精密称定,置具塞三角瓶中,精密加入稀乙醇20ml,称定重量,超声处理(120W,40KHz)30 分钟,放冷,称定重量,加稀乙醇补足减失的重量,摇匀,滤过,即得。
3.3 阴性样品溶液的制备
取除淫羊藿外的其余处方量药材,按制备工艺方法制备缺淫羊藿的阴性样品,按上述供试品溶液制备方法制成缺淫羊藿的阴性样品溶液。
3.4 对照品溶液的制备
精密称取淫羊藿苷对照品,加甲醇制成每1mL 含0.025mg的溶液,即得。
3.5 专属性考察
分别精密吸取对照品溶液、阴性样品溶液与供试品溶液各5μL,注入液相色谱仪,测定。结果表明,供试品溶液色谱中,在与淫羊藿苷对照品色谱峰相应位置上有相应峰,而缺淫羊藿阴性溶液色谱中无此峰,说明阴性无干扰,对照品、供试品、阴性样品色谱图分别见附图。
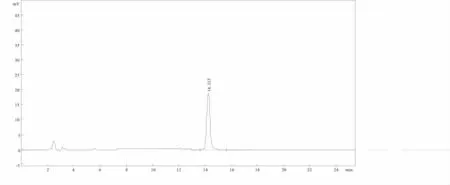
图4 淫羊藿苷对照品HPLC 色谱图

图5 前列癃闭通片HPLC 色谱图
图6 缺淫羊藿阴性样品HPLC 色谱图
3.6 线性关系考察
精密吸取浓度为0.1025mg/mL的淫羊藿苷对照品溶液(精密称取淫羊藿苷对照品10.25mg 置于100mL 量瓶中,加甲醇至刻度,摇匀,即得)2、4、6、8、10mL,分别置25mL 量瓶中,加甲醇稀释至刻度,摇匀,精密吸取10μL 进样,测定其峰面积积分值,以浓度为横坐标,峰面积积分值为纵坐标,绘制标准曲线,其回归方程为:A=881410.98×C-1173.3,r=0.9999。结果表明淫羊藿苷检测浓度在0.08~0.41μg 范围内与峰面积积分值具有良好的线性关系。
3.7 精密度试验
精密吸取同一份(批号20111125)供试品溶液10μl,重复进样5 次,测定。结果,RSD=0.86%(n=5),表明本方法精密度良好。
3.8 稳定性试验
分别精密吸取同一份(批号20111125)供试品溶液10μl,于0、3、6、9、12 小时进样,测定。结果,RSD=0.86%(n=5),表明供试品溶液在12 小时内基本稳定。
3.9 重复性试验
取同一批号样品(批号20111125)五份,按上述条件,平行处理并测定(n=2)。结果,RSD=0.95%(n=5),表明本方法重复性好。
3.10 回收率试验
精密称取已知含量为1.1373mg/g(批号20111125)样品约0.2g,共六份,分别精密加入淫羊藿苷对照品溶液(0.1025mg/mL)2mL,即0.2050mg,再加入稀乙醇18mL,按上述含量测定项下的方法测定含量,结果见表2。结果表明,本方法加样回收率好(样品进样量约为0.22μg,在线性范围内)。
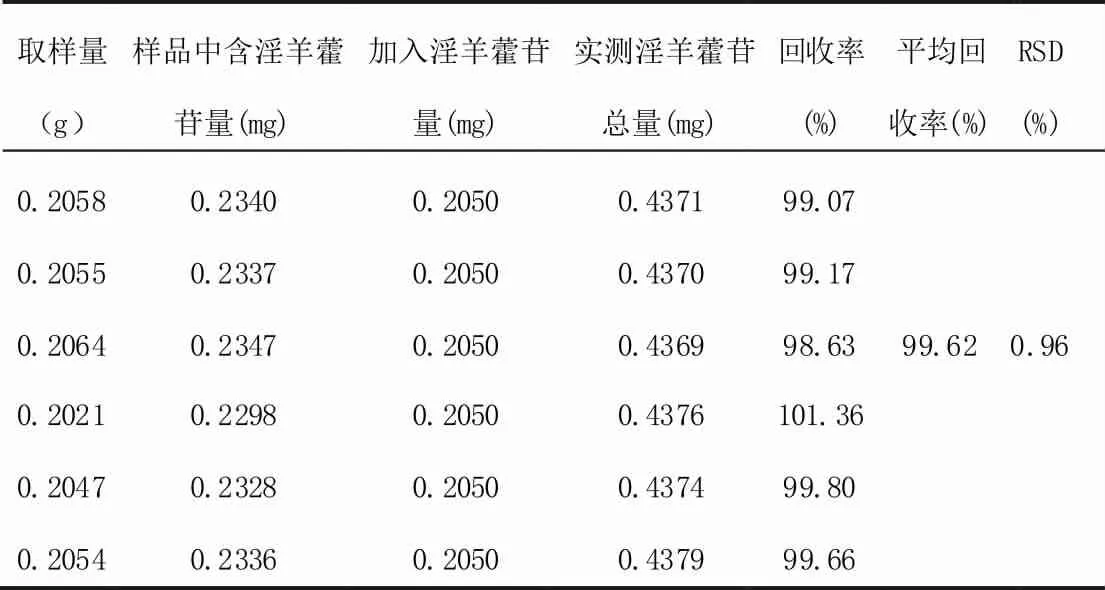
表2 回收率测定结果(n=6)
3.11 样品含量测定
取10 批样品,分别按"3.2"项下方法制成供试品溶液,照上述色谱条件测定,并计算淫羊藿苷的含量,结果见表3。
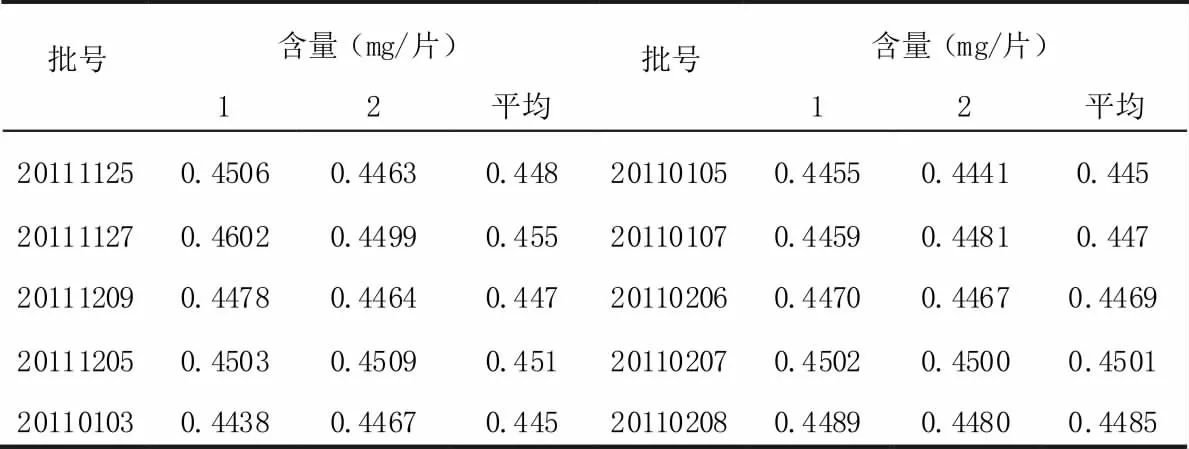
表3 样品含量测定结果(n=10)
4 讨论
供试品溶液的制备,本品用稀乙醇为提取溶媒进行超声提取,现对超声提取时间进行考察,结果表明:超声处理15 分钟时,样品中的淫羊藿苷提取不完全,超声处理30 分钟与45 分钟时,其含量相当,均可将样品中的淫羊藿苷提取完全,因此确定超声处理时间为30 分钟。参照中国药典2010年版一部淫羊藿项下淫羊藿苷的含量测定方法,对本品中淫羊藿苷的含量进行测定,试验结果表明,缺淫羊藿的阴性溶液无干扰,且样品分离效果好。经方法学考察,本含量测定方法结果较好,方法可行。经测定十批成品,片重0.4 克,其淫羊藿苷平均含量约为0.45 mg/片,按约80%折算,暂规定本品每片中含淫羊藿苷不得少于0.36mg。
[1]国家中成药标准汇编,内科肾系分册「S」.北京:人民卫生出版社,2002:876.
[2]刘克敏,刘振玉.黄芪的药理作用及其在运动医学中的应用「J」.现 代中 西 医 结 合 杂 志,2005,14 (17):2346-2347 -2349.
[3]陈营镑,刘峰.淫羊藿的研究进展「J」.陕西中医,2001,22(10):46.
[4]吕武清主编.中成药中的药材薄层色谱鉴别「M」.人民卫生出版社,1997:288,521.
[5]中华人民共和国药典(2010 版),一部「S」.2010 :附录32.
