淫羊藿中朝藿苷A、B和C的酶转化及其稀有箭藿苷的制备
2023-11-26彭磊徐丝瑜武博王政浩鱼红闪
彭磊,徐丝瑜,武博,王政浩,鱼红闪
(大连工业大学 生物工程学院,辽宁 大连,116034)
淫羊藿是小檗科(Berberdiaceae)淫羊藿属(Epimedium)多年生宿根性草本药用植物,又名仙灵脾,其茎、叶可入药,已有2 000多年的使用历史[1]。《本草纲目》中记载,其辛温无毒,具有坚筋骨、益精气、补腰膝、强心强骨等功能[2]。目前全世界淫羊藿属约有68个种,其中57个种生长在中国[3]。《中华人民共和国药典》2020年版第一部收载了4种药用淫羊藿,分别为:小蘖科淫羊藿(EpimediumbrevicornumMaxim)、箭叶淫羊藿[Epimediumsagittatum(Seib rt Zucc.) Maxim]、柔毛淫羊藿(EpimediumpubescensMaxim)和朝鲜淫羊藿(EpimediumkoreanumNakai)[4]。
淫羊藿的主要活性成分是黄酮类化合物,目前已从淫羊藿中发现了70余种黄酮类化合物,其中淫羊藿苷、朝藿苷A、B和C在淫羊藿中含量最多,它们分别占淫羊藿总黄酮含量的58.5%、5.92%、11%和24.5%,而其他黄酮类化合物含量很低[5]。常见的淫羊藿黄酮类化合物,如图1、表1所示。

表1 常见的淫羊藿黄酮类化合物Table 1 Common Epimedium flavonoids
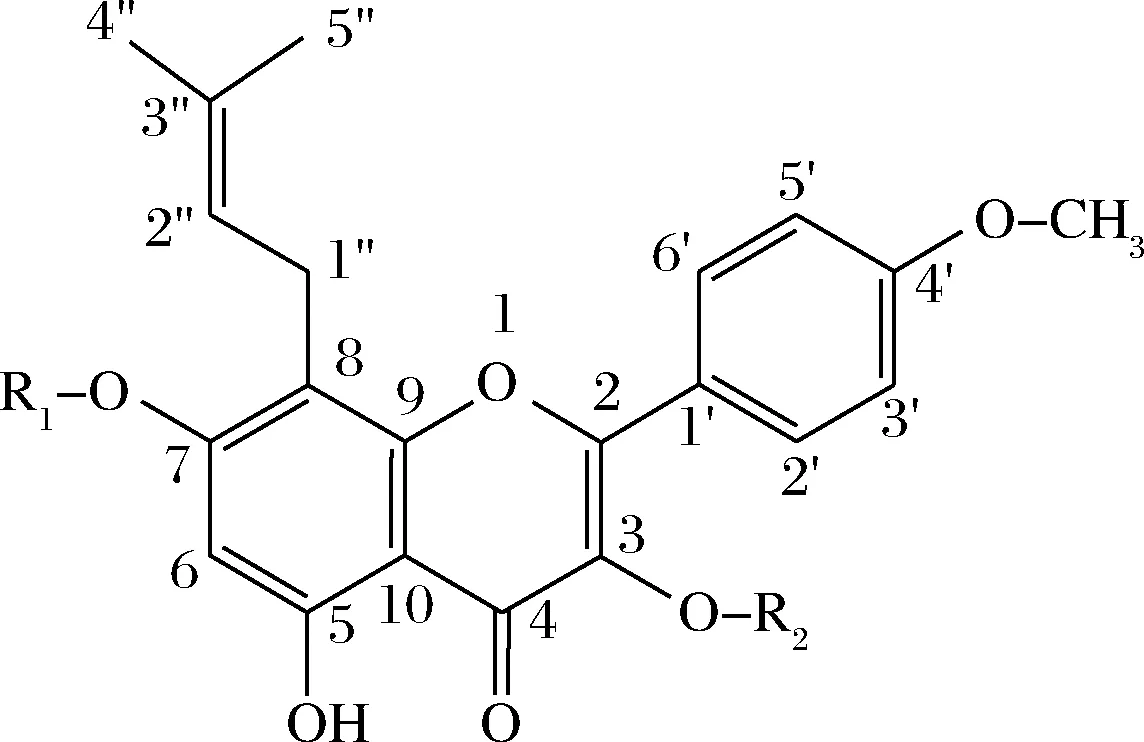
图1 常见的淫羊藿黄酮类化合物基本结构Fig.1 Basic structure of common Epimedium flavonoids
口服使用淫羊藿黄酮时,淫羊藿黄酮在肠道中被水解成低糖基黄酮苷或苷元,成为更容易吸收的化合物,通过肠道吸收最终起药效[6-7]。淫羊藿黄酮具有提高免疫、抗炎、抗衰老、抗肿瘤、抗骨质疏松、预防阿尔茨海默病和改善脑缺血的功能[8],淫羊藿苷元和淫羊藿次苷具有良好的抗癌、抗病毒、抗骨质疏松、抗炎、抗血栓和抗皮肤老化等功能且吸收率高[9]。现代科学研究表明,淫羊藿黄酮的药效与其糖基数量有关,糖基数量越少其吸收率和药效越高[10-11],但淫羊藿中含量高的朝藿苷A、B和C含有3个糖基,这导致其活性低、吸收率低。
因稀有淫羊藿黄酮类化合物产量少且价格昂贵,若用酶法水解淫羊藿中含量高的淫羊藿苷、朝藿苷A、B和C,制备箭藿苷A、B和C以及淫羊藿苷元等稀有淫羊藿黄酮类化合物,能为开发新药、保健食品和化妆品提供廉价的原料,有利于提高淫羊藿在中药产品、保健食品和化妆品中的利用率。
为此,本文利用Aspergillissp.y48所产的淫羊藿黄酮苷酶,水解淫羊藿中的朝藿苷A、B和C(淫羊藿苷制备后的废弃物)上的糖基,进而制备低糖基的箭藿苷A、B、C、淫羊藿次苷II和淫羊藿苷元,以期为淫羊藿苷A、B、C的高效利用和制备低糖基淫羊藿黄酮类化合物提供了理论基础和工艺方案。
1 材料与方法
1.1 材料
菌种Aspergillissp.y48,由本实验室从白酒生产曲类中分离得到[12]。硅胶板60-F254,由Merck公司生产。标准品:朝藿苷A、B、C;箭藿苷A、B、C;淫羊藿苷;淫羊藿次苷I;淫羊藿次苷II(宝藿苷I);淫羊藿苷元,由武汉格林特生物技术有限公司和南京广润生物制品有限公司生产。
1.2 酶的制备及活性确认
Aspergillissp.y48在100 mL含质量分数0.8%淫羊藿叶粉的5%麦汁培养基中,30 ℃下摇床培养6~7 d,离心分离菌体;向上清液中加入3倍体积的甲醇沉淀酶蛋白,静置过夜,离心收集酶蛋白沉淀;加入10 mL 0.02 mol/L(pH 5.0)的醋酸缓冲液溶解,离心去除不溶杂质,最终获得酶液。
酶活性确认:在试管中加入0.1 mL酶液与0.1 mL的质量分数0.2%的朝藿苷C,在45 ℃条件下反应1 h后,加入0.2 mL水饱和正丁醇终止反应;用正丁醇层的反应产物做薄层层析(thin layer chromatography,TLC),确认其所产酶的酶活性。
1.3 酶反应最适温度和最适pH的确定
最适温度:在试管中加入0.1 mL酶液与0.1 mL质量分数0.4%的朝藿苷C(最终为0.2%),分别在25、30、35、40、45、50、55、60、65、70 ℃下反应1 h,然后分别加入0.2 mL水饱和正丁醇终止反应,用正丁醇层做TLC分析,确认最适温度。
最适pH:分别在试管中加入0.1 mL pH 3.0、4.0、4.5、5.0、5.5、6.0、7.0、8.0质量分数0.4%的朝藿苷C溶液,分别与0.1 mL酶液混合,在45 ℃条件下反应1 h,然后分别加入0.2 mL水饱和正丁醇终止反应,用正丁醇层做TLC分析,确认最适pH。
1.4 朝藿苷A、B和C糖基的酶水解途径
在试管中,分别加入质量分数0.4%的0.1 mL朝藿苷A、B和C与0.1 mL酶液混合,在45 ℃下反应0.5、2、4、6 h,然后加入0.2 mL水饱和正丁醇终止反应,取朝藿苷A和C的0.5、4 h的正丁醇层以及朝藿苷B的0.5、6 h反应的正丁醇层做TLC分析,确认酶反应产物变化情况,进而确定酶水解朝藿苷A、B和C糖基的水解途径。
1.5 制备稀有淫羊藿黄酮类化合物箭藿苷A、B和C,以及淫羊藿苷元
箭藿苷A的制备:将40 mg朝藿苷A混合于10 mL的0.02 mol/L(pH 5.0)醋酸缓冲液,加入10 mL酶液,在45 ℃条件下反应3~4 h,加入20 mL水饱和正丁醇终止反应并萃取反应产物;再用10 mL水饱和正丁醇萃取2次。合并正丁醇,减压浓缩,干燥,得到箭藿苷A。
箭藿苷B的制备:将80 mg朝藿苷B混合于20 mL的0.02 mol/L(pH 5.0)醋酸缓冲液,加入20 mL酶液,在45 ℃条件下反应3 h,加入40 mL水饱和正丁醇终止反应并萃取反应产物;再用20 mL水饱和正丁醇萃取2次。合并正丁醇,减压浓缩,干燥,得到以箭藿苷B为主的产物。
箭藿苷C的制备:将400 mg朝藿苷C混合于80 mL的0.02 mol/L(pH 5.0)醋酸缓冲液,加入80 mL酶液,在45 ℃摇床反应3~4 h,加入50 mL水饱和正丁醇终止反应并萃取反应产物;再用40 mL水饱和正丁醇萃取2次。合并正丁醇,减压浓缩,干燥,得到箭藿苷C。
淫羊藿苷元的制备:将80 mg朝藿苷B混合于20 mL的0.02 mol/L(pH 5.0)醋酸缓冲液,加入20 mL酶液,在45 ℃条件下反应9 h,加入40 mL水饱和正丁醇终止反应并萃取反应产物;再用20 mL水饱和正丁醇萃取2次。合并正丁醇,减压浓缩,干燥,得到淫羊藿苷元。
1.6 薄层层析(thin layer chromatography,TLC)和HPLC法
TLC法:用反应后萃取的正丁醇,做TLC,在展开剂[V(乙酸乙酯)∶V(丁酮)∶V(甲醇)∶V(水)=8∶7∶1∶1]中展开,吹干,在紫外线280 nm下显色。用BandScan 5.0软件,确定TLC板的朝藿苷A、B和C反应液中底物、中间产物和最终产物斑点的含量比例,确定底物的酶转化情况。
HPLC法:将朝藿苷A、B和C反应后分离、干燥得到的反应产物,溶解于1 mL色谱纯甲醇,经0.45 μm滤膜过滤,做HPLC,确定朝藿苷A、B和C反应液底物、中间产物和最终产物的含量比例,以及底物的转化情况。HPLC色谱仪,Waters 2695高效液相色谱分析仪,Waters 2996二极管阵列检测器及Empower色谱工作站进行检测,色谱柱为中汇达Kromasil C18(250 mm×4.6 mm,5 μm)。进样量10 μL;柱温35 ℃;体积流速1.0 mL/min;检测波长273 nm;流动相:乙腈(A)-水(D)0~8 min,17% A~27% A;8~32 min,27% A等度;32~60 min,27% A~85% A;60~70 min,85% A等度;70~80 min,85% A~90% A。
2 结果与分析
2.1 酶反应最适温度与最适pH
按照1.2节及1.3节的方法进行操作,通过TLC检测分析朝藿苷C的生成量。如图2-a可见,随温度的变化,从20~45 ℃酶活性逐渐增高,45~70 ℃酶活性逐渐降低;如图2-b可见pH 3.0~5.0酶活性逐渐增高,pH 5.0~8.0酶活性逐渐降低。由此确定Aspergillissp.y48菌所产酶的酶反应最适温度为45 ℃,最适pH为5.0(图2)。
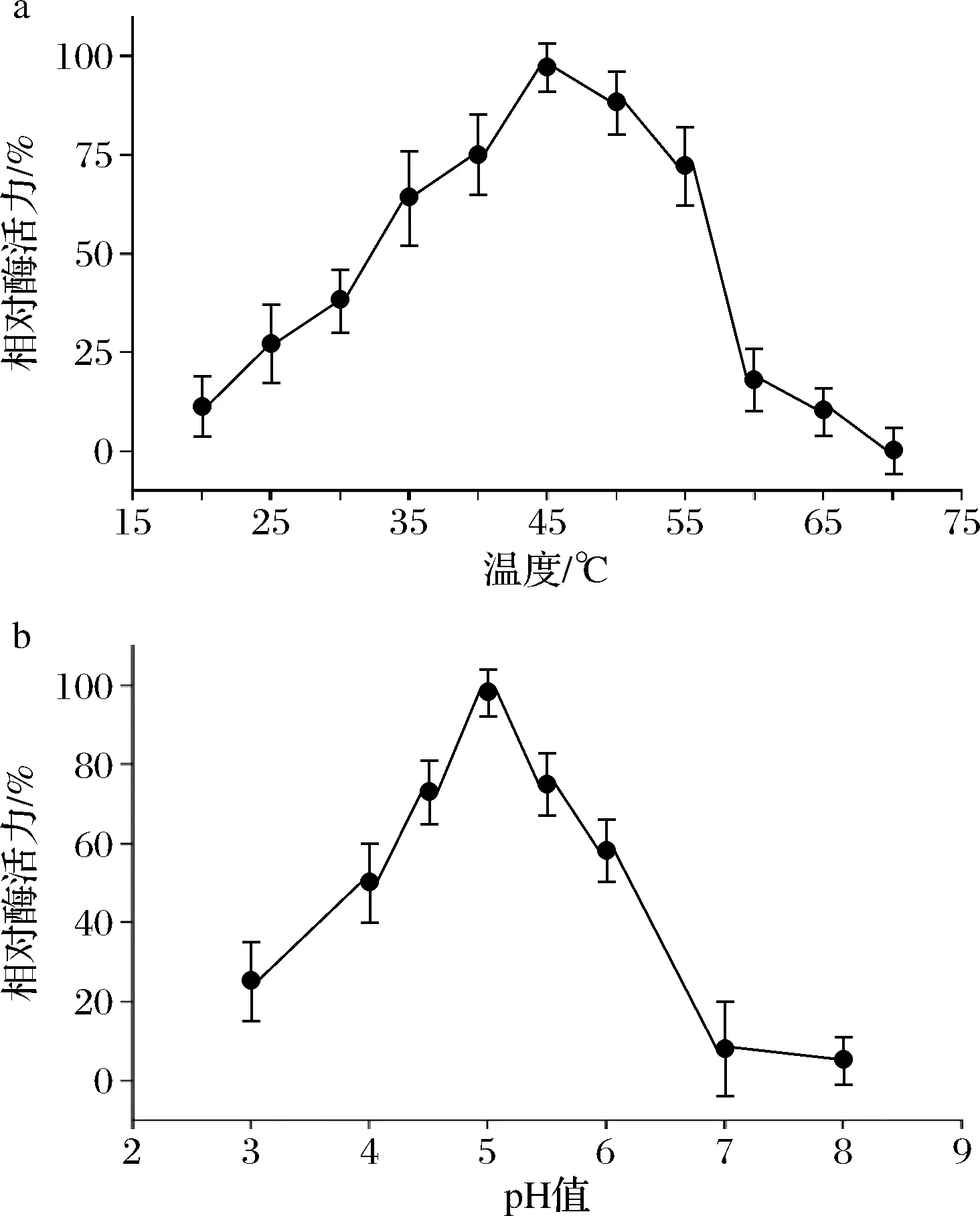
a-酶反应最适温度;b-酶反应最适pH图2 0.2%朝藿苷C在45 ℃酶反应1 h时最适温度和pHFig.2 Optimal temperature and pH of 0.2% epimedin C at 45 ℃ for 1 h by enzyme
2.2 朝藿苷A、B和C糖基的酶水解途径
采用1.4节的实验方法进行操作。取朝藿苷A和C的0.5、4 h的反应产物以及朝藿苷B的0.5、6 h反应的反应产物做TLC和HPLC分析,确认酶水解朝藿苷A、B和C糖基的水解过程,结果如图3所示。
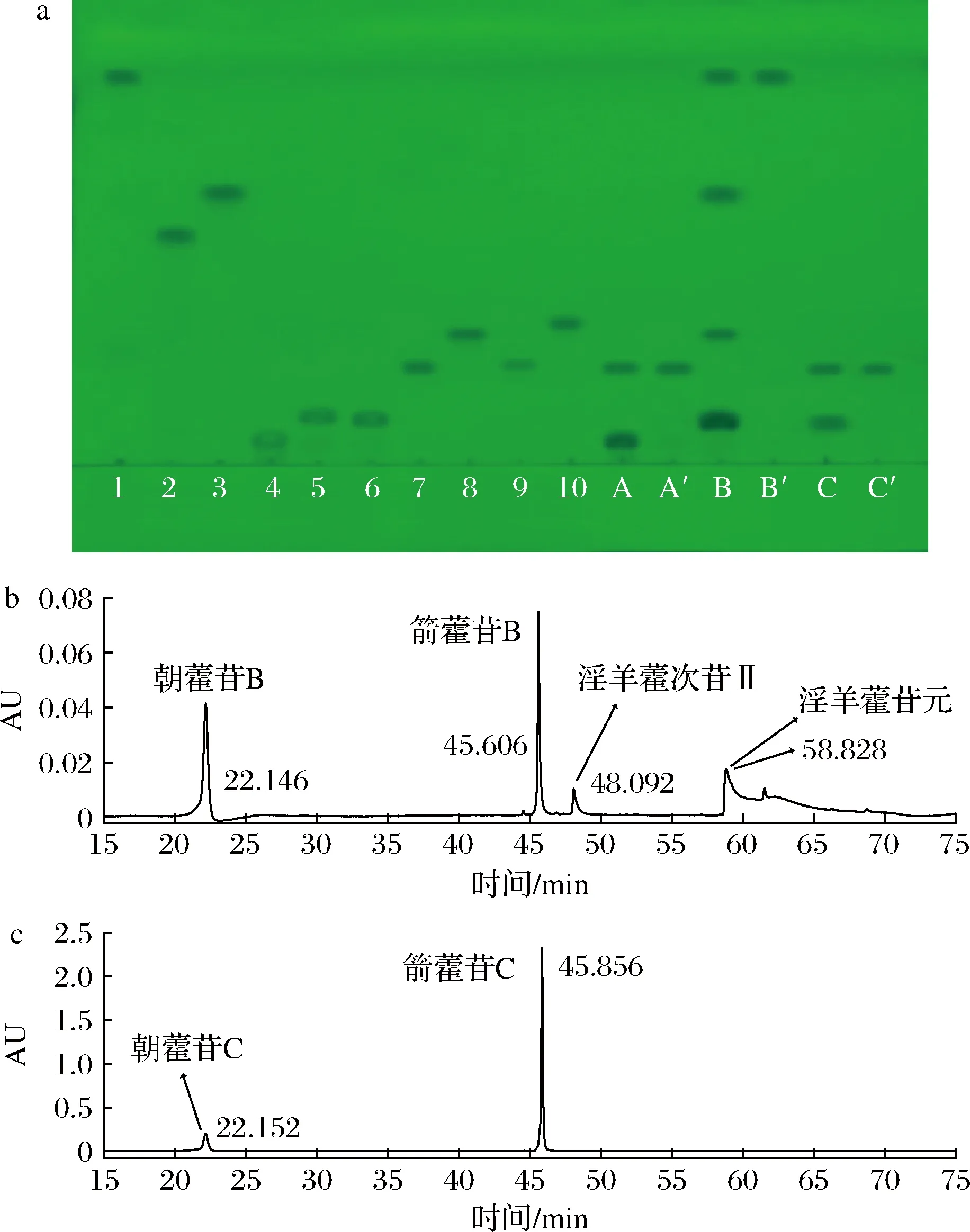
a-1-淫羊藿苷元;2-淫羊藿次苷Ⅰ;3-淫羊藿次苷Ⅱ;4-朝藿苷A;5-朝藿苷B;6-朝藿苷C;7-箭藿苷A;8-箭藿苷B;9-箭藿苷;10-淫羊藿苷标准品;A-朝藿苷A反应0.5 h;A′-朝藿苷A反应4 h;B-朝藿苷B反应0.5 h;B′-朝藿苷B反应6 h;C-朝藿苷C反应0.5 h;C′-朝藿苷C反应4 h;b-朝藿苷B反应0.5 h的HPLC 图;c-朝藿苷C反应0.5 h的HPLC图图3 0.2%朝藿苷A、B和C酶水解产物的TLC和HPLC图Fig.3 Enzyme reacting product from 0.2% epimedin A, B and C in TLC and HPLC
从图3中可见,朝藿苷A在45 ℃酶催化条件下反应0.5 h,朝藿苷A部分转化为箭藿苷A;反应4 h后,全部转化为箭藿苷A(图3-a的A和A′列);同样朝藿苷C在45 ℃酶催化条件下反应0.5 h,朝藿苷C部分转化为箭藿苷C;反应4 h后,全部转化为箭藿苷C(图3-a的C和C′行、图3-c);由此可见,Aspergillissp.y48所产酶能水解朝藿苷A和C的7-O-β-D-Glc糖基。
朝藿苷B在相同条件下反应0.5 h时,反应液中含有朝藿苷B、箭藿苷B、淫羊藿次苷II和苷元;当反应6 h后只有淫羊藿苷元(图3-a的B和B′列、图3-b)。由此可见,Aspergillissp.y48所产酶能水解朝藿苷B的7-O-β-D-Glc糖基,且可以水解箭藿苷B的末端3-O-β-D-Xyl(木糖基),变成淫羊藿次苷II;并进一步水解淫羊藿次苷II的3-O-α-L-Rha(鼠李糖基),转化成淫羊藿苷元。因此,朝藿苷A、B和C糖基的酶水解途径,如图4所示。
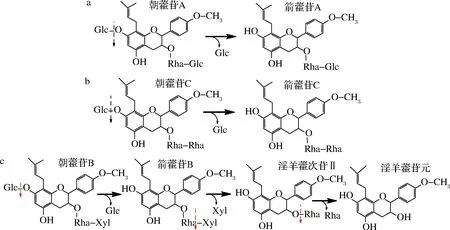
a-朝藿苷A;b-朝藿苷C;c-朝藿苷B图4 Aspergillis sp.y48菌酶水解朝藿苷A、B和C糖基的途径Fig.4 Hydrolysis pathways of epimedin A, B and C glycosides by enzyme from y48 strain
归纳上述结果,Aspergillissp.y48菌所产淫羊藿黄酮苷酶可以水解朝藿苷A、B和C的7-O-β-D-Glc糖基,转化生成箭藿苷A、B和C,通过控制酶反应时间可以将箭藿苷B末端3-O-β-D-Xyl(木糖基)转化成淫羊藿次苷II,并进一步水解淫羊藿次苷II的3-O-α-L-Rha(鼠李糖基)最终转化成淫羊藿苷元,因此该方法可以制备箭藿苷A、B、C和淫羊藿苷元。
2.3 从朝藿苷A、B和C酶转化制备淫羊藿稀有黄酮类化合物
上述实验证明了Aspergillissp.y48菌的淫羊藿黄酮苷酶能水解朝藿苷A和C的7-O-β-D-Glc糖基,可用于制备箭藿苷A和C。该酶能水解朝藿苷B的7-O-β-D-Glc糖基和3-O-糖基,生成箭藿苷B、淫羊藿次苷II和淫羊藿苷元。由此,控制朝藿苷B的酶反应时间,可制备箭藿苷B、淫羊藿次苷II和淫羊藿苷元。考虑到反应底物,朝藿苷A、B和C的水溶性,底物质量浓度定为2 mg/mL,进行将朝藿苷A、B和C用酶转化为稀有淫羊藿黄酮类化合物的实验。
根据1.5节的实验方法,制备箭藿苷A和C,得到61 mg HPLC纯度为98%的箭藿苷A(相对分子质量676.2、9.02 mmol),摩尔得率94.6%(图5-a);得到310 mg HPLC纯度为98%的箭藿苷C(相对分子质量660.7、469 mmol),摩尔得率96.5%(图5-b)。制备箭藿苷B和淫羊藿苷元:首先反应3 h,得到HPLC纯度为83%的箭藿苷B(相对分子质量646.6、89.7 mmol)58 mg,摩尔得率75.3%,其他为淫羊藿次苷II(图5-c);将反应产物中的箭藿苷B用制备色谱法分离,得到高纯度的箭藿苷B。将朝藿苷B的酶反应时间延长到6~8 h,使朝藿苷B全部转化为淫羊藿苷元;最终将80 mg朝藿苷B(相对分子质量808.8、98.9 mmol)酶转化制备为31 mg HPLC纯度为98%的淫羊藿苷元(相对分子质量368.4、84.1 mmol),摩尔得率93.8%(图5-d)。
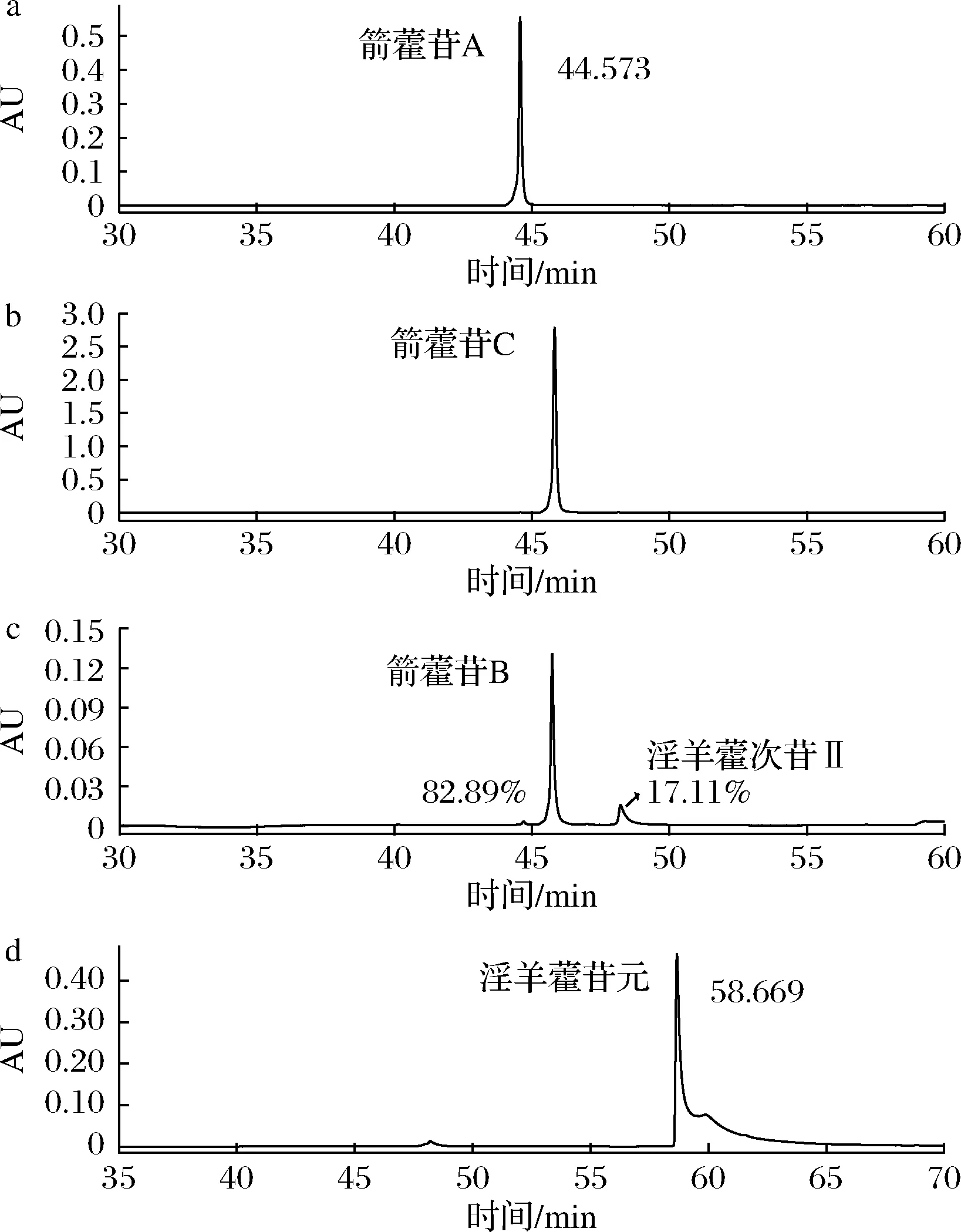
a-从朝藿苷A所制备的箭藿苷A;b-从朝藿苷C所制备的箭藿苷C;c-从朝藿苷B所制备的83%箭藿苷B;d-从朝藿苷B所制备的淫羊藿苷元图5 酶转化从朝藿苷A、C和B所制备的箭藿苷A、C和B的HPLC图Fig.5 Sagittatoside A, B and C products in HPLC fron epimedin C, A and B by enzyme
最终利用Aspergillissp.y48菌所产生的淫羊藿黄酮苷酶,将淫羊藿中含量高的朝藿苷A和C转化得到高纯度箭藿苷A和C,将朝藿苷B转化为箭藿苷B和淫羊藿苷元。该方法简单、成本低、收率高、产品纯度高,且迄今为止尚未见到其他相关报道。
3 结论与讨论
实验发现Aspergillissp.y48菌所产生的淫羊藿黄酮苷酶,能水解朝藿苷A、C和B的7-O-β-D-Glc糖基,分别生成箭藿苷A、C和B。其中该酶能进一步水解箭藿苷B的3-O-β-D-Xyl糖基,转化成淫羊藿次苷II,之后水解淫羊藿次苷II的3-O-α-L-Rha糖基,生成淫羊藿苷元,若延长反应时间,朝藿苷将全部转化为淫羊藿苷元。但该酶不能水解箭藿苷A的末端3-O-β-D-Glc糖基和箭藿苷C末端3-O-α-L-Rha糖基。该酶最适反应温度为45 ℃,最适pH为5.0。由此,该酶转化法可以用于利用朝藿苷A、C制备箭藿苷A、C,以及利用朝藿苷B制备箭藿苷B和淫羊藿苷元。
利用Aspergillissp.y48菌所产生的淫羊藿黄酮苷酶,将朝藿苷A和朝藿苷C转化为箭藿苷A和箭藿苷C的纯度均为98%,摩尔得率均94%以上;反应3 h能将朝藿苷B转化箭藿苷B,当酶反应时间延长到6~8 h时,朝藿苷B全部转化为HPLC纯度为98%的淫羊藿苷元,摩尔得率为93.8%。相比彭静等[13]报道的朝藿苷A和B的转化率73.69%和74.01%更高;相比杨轶舜等[14]报道的方法,本方法条件温和且得率更高。
相比之前的研究[15],本文对不同朝藿苷的酶转化进程进行了研究,分析了Aspergillissp.y48菌酶水解反应的时间,方法操作简单,收率高,成本低,产品易提取且纯度高,为高效利用淫羊藿黄酮类化合物和稀有淫羊藿黄酮类化合物的制备提供了方法参考。
