中国部分地区蝙蝠携带病毒的宏基因组学分析
2013-09-03杨凡力王意银郑文成何彪江廷磊李莹莹夏乐乐冯烨范泉水涂长春
杨凡力,王意银,郑文成,何彪,江廷磊,李莹莹,夏乐乐,冯烨,范泉水,涂长春
1 吉林大学畜牧兽医学院,吉林 长春 1300622 军事医学科学院军事兽医研究所,吉林 长春 1301223 成都军区疾病预防控制中心,昆明 云南 6501184 湖南省动物卫生监督所,长沙 湖南 4100075 东北师范大学吉林省动物资源保护与利用重点实验室,吉林 长春 130024
中国部分地区蝙蝠携带病毒的宏基因组学分析
杨凡力1,2,王意银3,郑文成4,何彪2,江廷磊5,李莹莹2,夏乐乐2,冯烨2,范泉水3,涂长春2
1 吉林大学畜牧兽医学院,吉林 长春 130062
2 军事医学科学院军事兽医研究所,吉林 长春 130122
3 成都军区疾病预防控制中心,昆明 云南 650118
4 湖南省动物卫生监督所,长沙 湖南 410007
5 东北师范大学吉林省动物资源保护与利用重点实验室,吉林 长春 130024
杨凡力, 王意银, 郑文成, 等. 中国部分地区蝙蝠携带病毒的宏基因组学分析. 生物工程学报, 2013, 29(5): 586−600.
Yang FL, Wang YY, Zheng WC, et al. Metagenomic analysis of bat virome in several regions of China. Chin J Biotech, 2013,29(5): 586−600.
蝙蝠携带有60多种病毒,其中许多对人有高度致病性。为了解中国蝙蝠携带病毒的自然本底、蝙蝠病毒的多样性和挖掘潜在的病毒病原,通过基于 Solexa高通量测序的病毒宏基因组学技术对从吉林、云南、湖南采集的蝙蝠组织进行病毒组学研究,获得了 11 644 232条读长 (Reads),并拼接出 44 872条重叠序列(Contig)。通过核酸序列注释发现,其中8.2% (4 002/44 872) 的重叠序列与病毒相关,能进一步注释到36个病毒科,包括19种脊椎动物病毒、6种植物病毒、4种昆虫病毒和4种噬菌体。通过对重叠序列的遗传进化分析、多序列比对显示,被注释为细小病毒、腺联病毒、博卡病毒、腺病毒、小双节RNA病毒等的重叠序列与已知病毒相似,部分序列却又呈现出明显的序列差异。通过对腺病毒和博卡病毒进一步的 PCR扩增证实了此研究方法可靠。旨在了解我国蝙蝠携带病毒组的构成,对建立高效的野生动物源人兽共患病的监测方法提供参考。
病毒宏基因组学,蝙蝠,病毒组,高通量测序
蝙蝠属于翼手目 (Chiroptera),是仅次于啮齿类的第二大类哺乳动物,占全球哺乳动物种类的20%,且分布广泛,除南北极外全球各个地区均有蝙蝠的存在[1]。近年来在蝙蝠体内发现的病毒逐渐增多,至 2007年为止,至少从蝙蝠体内检测或分离到 60多种病毒,其中包括一些致病性病毒,如埃博拉病毒 (Ebola virus)、亨德拉尼帕病毒 (Henipaviruses)、马尔堡病毒 (Marburg virus)、SARS冠状病毒 (SARS coronavirus)、狂犬病病毒 (Rabies virus) 以及狂犬病相关病毒(Lyssavirus) 等[2]。最近,Bokeloh和 Shimoni蝙蝠病毒、圆环病毒 (Circovirus)、博卡病毒(Bocavirus)、呼肠孤病毒 (Retrovirus)、星状病毒(Astrovirus) 和松湾病毒 (Cedar virus) 作为新病毒在蝙蝠体内发现[3-8]。2002年SARS冠状病毒疫情爆发,造成全球8 000多人感染,至少800人死亡[9]。研究表明我国的菊头蝠携带有 SRAS样冠状病毒 (SARS-like coronavirus),很可能是SARS样冠状病毒的自然宿主[10]。近期研究者又在加纳和欧洲的蝙蝠体内发现了与 2012年在中东地区引起2人死亡的人β冠状病毒2cEMC/2012(Human betacoronavirus 2cEMC/2012) 接近的 β冠状病毒[11]。因此,蝙蝠作为巨大的病毒贮存库越来越受到关注。我国自上世纪 80年代开展蝙蝠病毒研究以来,至少从蝙蝠体内检测或分离到16种病毒,包括基孔肯雅病毒 (Chikungunya virus)、罗斯河病毒 (Ross River virus)、乙型脑炎病毒 (Japanese encephalitis virus,JEV)、呼肠孤病毒和腺病毒 (Adenovirus)[6,12-15]。
我国蝙蝠有7科30属120种,分布广泛,多集中在华中、华南、西南等人口密集且气候湿润温热的地方[16]。因此应全面掌握我国蝙蝠携带病毒的自然本底以预防新发和再发人兽共患病。传统的病毒病原生态学研究方法,如细胞分离、核酸检测、血清学试验等都有一定的局限性。近年来基于高通量测序的病毒宏基因组学方法已经成为一个高效的工具用于对自然环境[17-18]、人[19]和动物[20-21]的病毒组学分析。目前采用病毒宏基因组学研究蝙蝠病毒在全球已有 4则报道[22-25],他们对蝙蝠粪便的病毒组研究在一定程度上反映了蝙蝠携带病毒的多样性,提供了有价值的数据,但目前对蝙蝠病毒的研究并不充分。为了建立蝙蝠病毒宏基因组学研究方法、丰富我国蝙蝠携带病毒的数据库,本研究自 2008年至2010年从我国三地采集到4种共241只蝙蝠样品,通过病毒宏基因组学对这些蝙蝠的肠道和肺脏组织进行病毒组学分析,发现了细小病毒(Parvovirus)、博卡病毒、小双节 RNA病毒(Picobirnavirus)、腺病毒等病毒的序列,描绘了上述蝙蝠组织样本携带病毒的情况,并通过对博卡病毒和腺病毒的 PCR验证,进一步证实了这些病毒的存在,为蝙蝠病毒的病原生态学研究提供一定的参考。
1 材料与方法
1.1 样品采集
2008年至2010年共采集了4种241只蝙蝠。2008年 12月在云南省靠近缅甸边境的拉波寨采集到 180只亚洲长翼蝠Miniopterus fuliginosus;2010年9月在吉林省长春市一座公寓顶层采集到31只萨氏伏翼Hypsugo savii和4只东方蝙蝠Vespertilio sinensis;2010年12月在湖南省慈利县采集到 26只大蹄蝠Hipposideros armiger。经形态学鉴定蝙蝠种类后立即解剖,将肠道 (含内容物)、肺脏分别取出装入2 mL冻存管,保存于−80 ℃。此研究严格按照军事医学科学院军事兽医研究所动物福利委员会相关规定执行。
1.2 试剂及材料
反 转 录 酶 (SuperScriptⅢ Reverse Transcriptase)、DNA 聚合酶 (AccuprimeTaqDNA Polymerase)、RNA酶抑制剂 (RNAseOUT)购自Invitrogen公司;核酸外切酶 (Exonuclease I)、碱性磷酸酶 (Shrimp alkaline phosphatase)、TRIzol (Total RNA 提取试剂) 购自宝生物 (大连) 公司;QIAquick PCR Purification Kit购自QIAGEN 公司;DNA 酶 (Turbo DNase) 购自Ambion公司;核酸酶 (Benzonase Nuclease) 购自Novagen公司;DNA聚合酶Klenow片段购自NEB公司;0.22 μm针头滤器和PelliconⅡ膜包购自Millipore公司;SM缓冲液 (50 mmol/L Tris,10 mmol/L MgSO4,0.1 mol/L NaCl,pH 7.5);DNA提取试剂盒购自 QIAGEN公司;PCR反应液购自天根生物公司。
1.3 样品制备
在生物二级安全柜内将吉林采集的 35只蝙蝠和湖南采集的 26只蝙蝠的肺脏和肠道样本每只各取大约0.1 g,云南采集的180只蝙蝠随机挑选出 40只蝙蝠的肺脏和肠道各取大约 0.1 g混合。不同采集地点混合成1组,共3组 (云南组名称为 pool-1;湖南组为 pool-2;吉林组为pool-3)。每一个组按1:10 (W/V) 加入无菌SM缓冲液,用 Waring组织研磨仪磨碎;研磨液于4 ℃、10 000 r/min离心30 min;上清液先后用0.45 μm和0.22 μm孔径的PelliconⅡ膜包滤掉组织碎片、细菌和其他杂质;滤液使用截留分子量为100 kDa的PelliconⅡ浓缩膜包进行浓缩;浓缩液用 Beckman 超速离心机 SW55Ti转头45 000 r/min离心2 h;沉淀用SM缓冲液悬浮并使用0.22 μm针头滤器过滤;为降低游离核酸污染,每 116 μL的滤过液添加 14 U的 DNA酶(Turbo DNase)、25 U 的核酸酶 (Benzonase Nuclease)、20 U的核酸外切酶 (RNase I) 以及10×Turbo DNase缓冲液在37 ℃消化1 h;随后用TRIzol提取核酸 (包括RNA和DNA),并溶解于20 μL的无RNA酶的水中。
1.4 反转录及随机PCR
每组分别将8 μL上述病毒核酸加入1 μL二甲基亚砜 (DMSO) 和 1 μL 50 μmol/L 的带有20 bp锚定序列的6mer随机引物 (pool-1: 5¢-GC CGGAGCTCTGCAGATATCNNNNNN-3¢;pool-2:5¢-GTATCGCTGGACACTGGACCNNNNNN-3¢;pool-3: 5¢-CGCATTGGTCGGCACTTGGTNNNN NN-3¢)。于 72 ℃作用 5 min,然后立即冰浴 2 min;加入 1 μL DTT、1 μL dNTP mixture (10 mmol/L)、20 U RNAseOUT、4 μL 5×缓冲液、200 U Superscript Ⅲ和 ddH2O,25 ℃反应 10 min,50 ℃反应50 min合成cDNA,85 ℃ 1 0 min 灭活反转录酶。加入5 U DNA聚合酶Klenow片段37 ℃反应60 min合成双链cDNA并75 ℃灭活10 min,随后在此体系中加入 2 U碱性磷酸酶 (Shrimp alkaline phosphatase) 和 2.5 U 核酸外切酶(Exonuclease I) 于37 ℃反应60 min,除去多余的引物和游离的核苷酸,并75 ℃灭活10 min。然后取 10 μL 上述模板,加入 2 μL 10 μmol/L 锚定序列引物 (pool-1: 5¢-GCCGGAGCTCTGCAGAT ATC-3¢;pool-2: 5¢-GTATCGCTGGACACTGGA CC-3¢;pool-3: 5¢-CGCATTGGTCGGCACTTGG T-3¢)、5 μL 10×AccuPrime bufferⅠ、1 μL DNA 聚合酶 (AccuprimeTaqDNA Polymerase) 及ddH2O,共50 μL体系进行不依赖序列的单引物扩增 (Sequence-independent single primer amplification,SISPA):94 ℃ 30 s,55 ℃ 30 s,68 ℃ 1 min ,35个循环。扩增产物使用QIAquick PCR Purification Kit试剂盒纯化并溶于50 μL的TE缓冲液中。
1.5 Solexa高通量测序
纯化后的PCR产物送华大基因公司 (深圳)进行Solexa高通量测序。将3组样品混合后使用超声法打断到180 bp左右,末端分别加上接头,通过接头 PCR将 DNA片段连接到芯片上形成“桥”(Bridge) 并制成文库,随后应用4种荧光标记的核苷酸通过桥式PCR (Bridge PCR) 进行边合成边测序 (Sequencing By Synthesis,SBS),获得原始读长数据,去掉长度<100 bp的读长和锚定引物序列,通过SOAPdenove软件拼接成重叠序列 (Contig),随后进行数据库比对分析。首先将获得的重叠序列使用 BLASTn和 BLASTx在 GenBank的非冗余核苷酸序列数据库(Nonredundant database) 中进行比对,将E value <10−3的序列作为有意义的序列,然后按照注释到的物种信息去掉细菌及真核类序列,余下的病毒相关序列通过系统进化分析和多序列比对进行鉴定。
1.6 病毒样序列分析
根据BLAST注释信息,挑选E value较小且同源性较高的病毒样重叠序列使用 DNAStar编辑,整理后用 MEGA5.0[26]软件进行系统发生分析,进化树选择基于 Maximum Composite Likelihood算法的邻接法 (Neighbor-joining method,NJ),Bootstrap检验设定为1 000个重复。使用DNAstar软件包的MegAlign软件对部分低同源性的病毒样序列和 GenBank中参考序列的氨基酸序列进行多序列比对,从而进行人工辅助鉴定。
1.7 PCR检测验证博卡病毒和腺病毒
使用博卡病毒和腺病毒序列信息,分别设计了套式 PCR对这两种病毒进行验证。通过GenBank下载博卡病毒参考序列,使用 Primer Premier 5.0软件设计检测博卡病毒的简并引物。腺病毒的检测引物参考已发表的文献[15]。使用QIAGEN的DNA提取试剂盒对保存的蝙蝠组织样品 (肠道和肺脏) 单样提取 DNA,DNA提取后进行PCR检测。PCR反应体系为:PCR反应液 (天根) 25 μL、ddH2O 19 μL、上游引物(10 pmol/μL) 2 μL、下游引物 (10 pmol/μL) 2 μL、DNA模板2 μL。PCR温度条件为:94 ℃预变性2 min;然后94 30 s℃、53 30 s℃、72 40 s℃,外套反应和内套反应各 35个循环;72 ℃延伸7 min。PCR产物经电泳鉴定为阳性后,送吉林库美基因公司测序。将获得的序列构建系统进化树,方法与重叠序列建树方法一致。
2 结果
2.1 测序DNA样品的制备
经过病毒纯化、核酸提取后使用ND1000核酸蛋白紫外定量检测仪对3组样品的核酸浓度进行定量。所提取 RNA和 DNA总浓度均高于200 ng/μL。进过反转录和不依赖序列的单引物扩增,产物条带为抹带,分布于2 000 bp以内,主要集中在100 bp至500 bp的区域 (图1)。3组的PCR产物经纯化回收后经ND1000核酸蛋白紫外定量检测仪测定 260/280吸光值比在 1.8至 2.0之间,样品浓度>800 ng/μL。符合Solexa测序的要求。
2.2 Solexa测序结果
图1 不依赖序列的单引物扩增结果Fig. 1 Sequence-independent single primer amplification results of viral nucleic acids. M: DNA marker.
通过高通量测序,获得了11 644 232个读长,并进一步拼接成48 872个重叠序列,平均长度为136.7 bp。与NCBI病毒数据库比对,4 002条重叠序列注释为病毒,占所有重叠序列的 8.2%,其中2 310条可注释到36个不同的病毒科,另1 692条重叠序列被定义为疑似病毒序列但未被分类。在这36个科中,44.6% (1 030/2 310) 的序列与 19个脊椎动物病毒科相关,包括腺病毒科 (Adenoviridae)、疱疹病毒科 (Herpesviridae)、乳头瘤病毒科 (Papillomaviridae)、细小病毒科(Parvoviridae)、圆环病毒科 (Circoviridae)、冠状病毒科 (Coronaviridae)、小双节 RNA 病毒科(Picobirnaviridae)、 小 RNA 病 毒 科(Picornaviridae)等;昆虫病毒相关序列占到了6.9% (160/2310),包括囊泡病毒科(Ascoviridae)、杆状病毒科 (Baculoviridae)、多DNA病毒科 (Polydaviridae)、布尼亚病毒科(Bunyaviridae) 4个科;植物病毒序列占 1.8%(41/2310) 包括花椰菜花叶病毒科(Caulimoviridae) 等 6个科;另外,还有43.0%(993/2310) 的序列与噬菌体相关,包括长尾噬菌体科 (Siphoviridae)、短尾噬菌体科(Podoviridae) 等4个科 (图2)。在脊椎动物病毒序列中,腺病毒、圆环病毒、腺联病毒(Adeno-associated viruses,AAV)、博卡病毒、细小病毒、小双节RNA病毒的序列在GenBank中能找到较好的比对结果,即 E value<10−20,比对得分>100,且核苷酸或氨基酸序列与已知序列同源性在 60%至 98%,经遗传进化分析和分子生物学鉴定后可以确认为已知病毒和新发现的病毒序列;而疱疹病毒、乳头瘤病毒、冠状病毒等相关序列的很大一部分无论是核酸序列还是氨基酸序列与现有序列同源性低于 60%,且E value在 10−2~10−5之间,目前可将其定义为新病毒相关序列。

图2 Solexa高通量测序获得的重叠序列注释Fig. 2 Annotation of Solexa contigs. The numbers and percentages of each virus species were labeld on left. The figure on right is the number of the contigs, which are classified into 36 viral families.
2.3 细小病毒 (Parvovirus,PV) 重叠序列鉴定
本研究有 2条重叠序列注释到细小病毒属(Parvovirus)。其中一条为291 nt的NS1基因片段。通过与GenBank数据库BLASTx比对,发现与之同源性最高的为小鼠微小病毒 (Mice minute virus),局部有70%的氨基酸同源性;另一条重叠序列为114 nt的VP1基因片段,与小鼠微小病毒有74%的氨基酸序列同源性。使用相对较长的 291 nt的重叠序列 (BtPV-0866) 构建的系统进化树。选取细小病毒属的代表序列作为参考序列的同时用腺联病毒 2型 (AAV-2) 作为外群对照。进化树显示BtPV-0866处在细小病毒属的外围,形成一个独立分支 (图 3A)。根据核苷酸序列推导的氨基酸序列长度为95个残基,与小鼠微小病毒该区域氨基酸同源性为56% (图3B)。利用氨基酸序列进行多序列比对发现,BtPV-0866在保守基序处与参考序列完全相同,如N端第6~13位的DQKGKGSK、第31~48位仅有2个残基的区别,在第66~80位区域变异较大,与参考序列仅有一个残基相同;而作为外群对照的 AAV-2氨基酸序列仅在少数位点与细小病毒属基序相同。根据核苷酸序列和氨基酸序列的鉴定,认为所获得的重叠序列来自新的细小病毒。
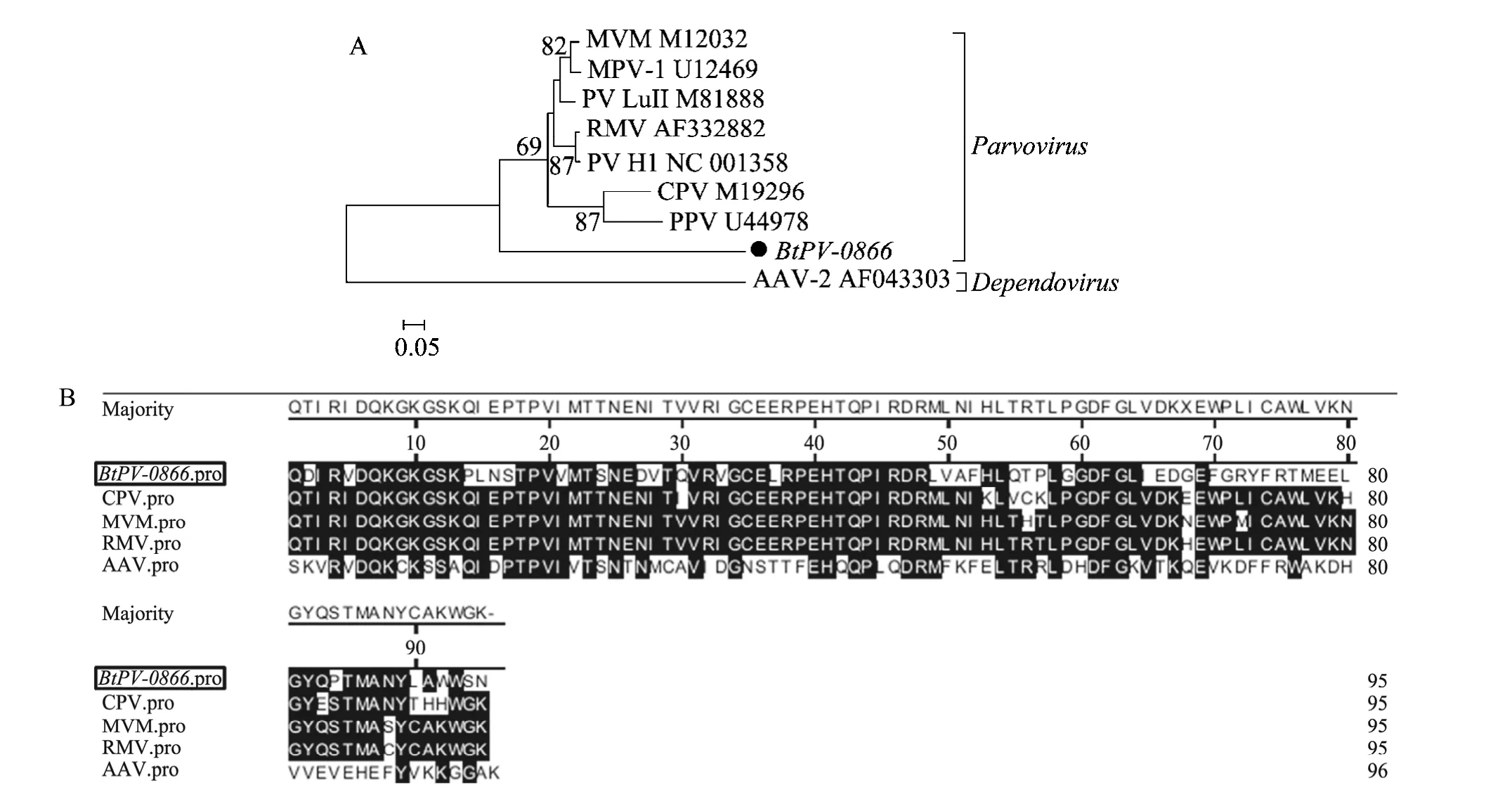
图3 细小病毒重叠序列鉴定Fig. 3 Characterization of PV-like contigs. (A) Phylogenetical analysis of PV-like contig BtPV-0866 with reference sequences based on about 290 nt NS1 gene. (B) Multiple sequences alignment of deduced amino acid sequences of parvovirus NS1. The black boxed aa sequence is majority sequence.
2.4 腺联病毒 (Adeno-associated viruses,AAV) 重叠序列鉴定
之前的研究已在棕果蝠、菊头蝠、鼠耳蝠等蝙蝠粪便内检测到AAV。本研究中共11条重叠序列被注释到依赖病毒属 (Dependovirus),其中7条 (长度在108~318 nt之间) 与在云南省的大足鼠耳蝠粪便中发现的蝙蝠腺联病毒 YNM 株(BtAAV-YNM)[27]在6个不同的区域有78%~95%的核苷酸同源性 (图4A)。如168 nt的重叠序列BtAAV-0854与BtAAV-YNM的Rep基因具有高达95%的核苷酸同源性,而192 nt的BtAAV-2413与BtAAV-YNM的Cap基因具有高达90%的核苷酸同源性。其余4条重叠序列与GenBank数据库中的AAV序列有46%~60%的核苷酸同源性,说明还存在其他与已知的 AAV遗传距离较大的病毒。对裁剪后 150 nt的Cap基因序列BtAAV-2143和BtAAV-3759的遗传进化分析显示BtAAV-2143与BtAdV-YNM处于同一分支,遗传距离较近;而BtAAV-3759独立成为一个分支,与AAV代表株遗传距离较大,属于依赖病毒属一个新的种 (图4B)。综合对11条疑似AAV重叠序列的分析,虽无法确定这 11条重叠序列来自多少株病毒,但可以确定至少有 1株与BtAAV-YNM同源性较高的AAV和1株与已知的病毒株变异较大的AAV存在这批蝙蝠样本中。

图4 腺联病毒重叠序列鉴定Fig. 4 Characterization of AAV-like contigs. (A) Sequence comparison of AAV-like contigs with BtAAV-YNM. The percentages above bars are similarity of contigs with BtAAV-YNM. (B) Phylogenetical analysis of AAV-like contigs with reference sequences based on 150 nt Cap gene.
2.5 小双节RNA病毒 (Picobirnavirus,PBV)重叠序列鉴定
本研究获得的3条PBV相关的重叠序列,1条位于第一节段与兔小双节 RNA病毒 (Rabbit picobirnavirus) 具有50%的核苷酸同源性。另外两条位于第二节段,与人小双节 RNA病毒(Human PBV GPBV11) 分别有61%和63%的核苷酸同源性。通过重叠序列BtPBV-1798与其他来源的PBV第二节段 (编码RdRp) 303 bp局部序列的进化分析显示出与现有的 PBV中的Human PBV GPBV11相对距离较近,处于一个进化分支上 (图5A)。将核苷酸序列翻译为97个残基,与Human PBV GPBV11同源性为55%。多序列比对显示该残基序列与参考序列具有共同的保守残基,如位于序列 N端的 QEGGP、DDVKQR和序列中部的AVD、FYQP、TDFTK、HFN等保守基序。而在氨基酸序列变异较大的区域,如37至49位和第84至96位残基,重叠序列以及其他参考序列相互之间均有较大的变异(图 5B)。

图5 小双节RNA病毒重叠序列鉴定Fig. 5 Characterization of PBV-like contig. (A) Phylegenetical analysis of PBV-like contigs with reference sequences based on 303 nt RdRp gene. (B) Multiple sequence alignment of deduced amino acid sequences of PBV. The black boxed aa sequence is majority sequence. The motif QEGGP, DDVKQR, FYQP, TDFTK and HFN are conservative.
2.6 博卡病毒 (bocavirus,BoCV) 重叠序列鉴定及PCR验证
本研究发现了一条重叠序列注释到细小病毒科的博卡病毒属 (Bocavirus)。BLAST比对发现这条序列位于博卡病毒NP1基因,与 CnMV核苷酸同源性最高,为62%。通过PCR扩增博卡病毒588 nt的VP1部分片段,在云南的亚洲长翼蝠肠道样品中检测出 2份阳性样品 (图 6A)。经测序两样品之间同源性100%,为同一毒株,暂命名为 Bat bocavirus YNMi-1 (GenBank Accession No. KC172378)。该序列与 CnMV有55%的氨基酸同源性。PCR扩增序列与参考序列进行系统进化分析显示所获得的 Bat bocavirus YNMi-1序列与CnMV、海狮博卡病毒 (Sea lion BoCV)、猫博卡病毒 (FBoCV) 处于同一个进化分支,但在这个进化分支上 Bat bocavirus YNMi-1和另外3株病毒有相对较大进化距离。YNMi-1与此前在海南鼠耳蝠体内发现的1株蝙蝠博卡病毒 (Myotis BoCV-1) 处在不同的进化分支且仅有56%的核苷酸同源性 (图6B),说明两株蝙蝠博卡病毒无直接的进化关系即蝙蝠博卡病毒具有基因多样性。利用翻译的氨基酸序列与参考序列进行多序列比对显示,本研究所扩增的 DNA片段序列中部在标尺的第 46~68位(HVR1)、84~133 位 (HVR2)、179~194位 (HVR3)存在3个高度变异区,序列之间无论是氨基酸的种类还是长度都有很大差异。YNMi-1在 HVR1区域相比Sea lion BoCV和FBoCV缺失了6个氨基酸残基;在HVR2和HVR3区域,YNMi-1相对于主要序列分别额外插入了8个和5个氨基酸残基 (图6C)。YNMi-1在中部的保守基序如标尺标记的69~82位与主序列仅有1个残基的差别,在137~147位残基完全相同。
2.7 腺病毒 (Adenovirus,AdV) 重叠序列鉴定及PCR验证
本研究有 69条重叠序列注释为哺乳动物腺病毒属 (Mastadenovirus)。BLAST比对发现这些序列都属于哺乳动物腺病毒的hexon、polymerase、penton、pX、pVI等基因。其中3条重叠序列与 GenBank中西班牙的一株腺病毒Has070613-2pol基因或hexon基因有分别高达98%,96%和91%的核苷酸同源性。体现了同一蝙蝠腺病毒毒株分布的广泛性。其余重叠序列与已知的腺病毒序列同源性均低于83%。
通过套式PCR扩增腺病毒270 ntpol基因检测,从湖南的样品中检测出一份腺病毒阳性样品(图 7A),暂命名为 bat adenovirus HNHi-1(GenBank Accession No. KC171020)。经比对与犬腺病毒 (CAdV)和猪腺病毒 A (PAdV-A) 具有71%的核苷酸同源性,与蝙蝠腺病毒 A(BtAdV-A) 和蝙蝠腺病毒 B (BtAdV-B) 具有70%的核苷酸同源性。系统进化分析显示BtAdV-HNHi-1属于哺乳动物腺病毒与 CAdV、BtAdV-A、BtAdV-B、PAdV-A处在同一个大的分支,相对与犬腺病毒距离最近 (图 7B)。由于扩增片段较短,新发现的蝙蝠腺病毒与其他蝙蝠腺病毒的进化关系还有待进一步研究。

图6 博卡病毒PCR扩增和序列鉴定Fig. 6 Characterization of bocavirus amplicon. (A) The result of bocavirus PCR amplification. M: DNA marker DL2000; lane 1 to 19 represent samples; lane 20: ddH2O. (B) About 585 nt VP1 gene-based phylogenetical analysis of bocavirus PCR amplicon with reference. (C) Multiple sequences alignment of deduced amino acid sequences of bocavirus. The black boxed aa sequence is majority sequence.

图7 腺病毒PCR扩增和序列鉴定Fig. 7 Characterization of adenovirus amplicon. (A) The result of adenovirus PCR amplification. (B) Phylegenetical analysis of adenovirus PCR amplicon compared with reference sequences based on about 270 nt hexon gene.
3 讨论
随着人类活动区域的不断扩大,蝙蝠的生存空间遭到侵占,创造了更多蝙蝠与人类接触的机会,使得许多高致病性的病毒由蝙蝠传播给人类或家畜。发现蝙蝠携带的高致病性病毒和新型病毒、监控蝙蝠病毒变异,已成为人们进行蝙蝠病毒流行病学研究的重要内容。病毒宏基因组学技术在这一领域的研究中效率最高、覆盖的病毒面最广。目前病毒宏基因组学不仅已用在水体、土壤等无机环境,也在人、浣熊、火鸡、海龟等动物携带病毒的研究中展现出明显优势,其获得的结果系统地描绘了它们的病毒组[28]。本研究通过建立组织病毒宏基因组学方法,对蝙蝠的呼吸系统和消化系统 (包括内容物) 两类组织进行病毒组学研究。通过Solxa测序和生物信息学注释获得的病毒相关重叠序列占总数的8.2%,而之前的病毒宏基因组研究病毒序列占总序列数的0.1%~58%不等[22-25]。并且注释为病毒的丰度与前面4份蝙蝠病毒宏基因组研究结果也有差异,首先在本研究中噬菌体占了43.0%,属于仅次于脊椎动物病毒的第二大类的病毒,在以前美国和我国的一例研究结果显示噬菌体的序列在多个混样 (pool) 中数量居第一或第二[22,24];其次,脊椎动物病毒所占比例最大,高达44.6%,而之前美国和我国的蝙蝠粪便病毒组研究结果显示的脊椎动物病毒含量相对较少,甚至低于10%。而本研究中的昆虫病毒和植物病毒序列数量较少,而在之前的中国蝙蝠粪便的病毒组研究中昆虫病毒占第二位置[24]。这些区别可能是因为我们所处理的肠道和肺脏组织相对较多而粪便含量少,而之前的研究集中在蝙蝠排出的粪便,而昆虫病毒和植物病毒恰恰只存在于蝙蝠的粪便中,并不在蝙蝠体内细胞中增殖。
本研究尝试利用加在随机引物前端的锚定引物序列的差异对湖南、云南、吉林三地的蝙蝠病毒进行分组,但由于在测序时使用超声法将DNA打断到 180 bp左右,我们所扩增的DNA产物长度大于500 bp的DNA片段被打断后,随机 PCR产物中间部分序列丢失了锚定引物序列信息,无法完成分组,故本次高通量测序结果做数据处理时放弃了对蝙蝠病毒进行地域上的分组。但病毒株的地理信息我们仍然可以从后续的PCR验证中获得。在以后的研究中,可以通过凝胶回收300 bp以内的随机PCR产物进行高通量测序来解决这一问题,从而对不同组样品的测序结果进行区分。
疱疹病毒 (Herpesvirues)、乳头瘤病毒(Papillomavirues) 等相关序列与已知病毒有着低于60%甚至更低的同源性,这些低同源性的序列可能源自一些我们尚不知道的病毒,相信随着病毒数据库的不断完善,这些类似病毒序列将被注释到。还有其他一些重叠序列,如牛病毒性腹泻病病毒 (BVDV),所获得重叠序列虽然与BVDV具有高达90%以上的同源性,但同时与动物染色体上的一段基因序列也有着很高的同源性。这可能反映了病毒基因能整合到某些宿主基因组中,这也是Cui等能从蝙蝠脑组织的转录组中发现反转录病毒的原因[29]。
本研究发现的病毒包括细小病毒、博卡病毒、小双节RNA病毒、腺病毒和腺联病毒等。博卡病毒属于细小病毒亚科,博卡病毒属,目前国际病毒分类命名委员会 (ICTV) 规定该属仅有两个种:牛细小病毒 (Bovine parvovirus,BPV)和犬微小病毒 (Canine minute virus,CnMV)[30],近年来博卡病毒的宿主范围从牛、犬扩大到了人、猩猩、猪、海狮、蝙蝠等动物。人博卡病毒能造成儿童的呼吸道感染[31]、胃肠炎和腹泻[32];猪博卡病毒则在呼吸道感染的仔猪体内有较高阳性率[33],因此该病毒具有公共卫生意义。小双节 RNA病毒科目前只有小双节 RNA病毒属(Picobirnavirus),成员为具有两个节段的双链RNA病毒,人的小双节RNA病毒常在腹泻病人的粪便中发现,在家畜、禽类、自然环境中也能检测到 PBV[34]。腺病毒科包含 5个属,均为无囊膜的双链DNA病毒,感染谱广泛,能在几乎所有种类的脊椎动物体内发现[35]。其中人腺病毒的一些血清型能造成呼吸道炎症和流行性结膜炎[36-37]。本研究通过建立组织病毒宏基因组学方法,对我国三地蝙蝠组织病毒组进行研究,并对一部分结果进行了系统进化分析、多序列比对以及 PCR检验。其中一些序列无论核苷酸序列的同源性、序列的遗传距离还是推导氨基酸序列的特征都支持这些序列可能是新病毒,如细小病毒、博卡病毒、腺病毒、小双节RNA病毒、圆环病毒等。还有一些序列与已知病毒序列有高于90%的同源性,如腺联病毒、辣椒温和斑点病毒、部分腺病毒以及多种噬菌体等。本研究未发现该地区蝙蝠携带有埃博拉病毒、尼帕病毒、SARS冠状病毒等已知对人具有高致病性的病毒,但我们不应放松对新发和再发蝙蝠源人兽共患病的监测。我国蝙蝠分布广泛、种类众多,全面地研究我国不同地区的蝙蝠病毒组为防控蝙蝠将病毒传播给人类提供了重要信息。
[1]Teeling EC, Springer MS, Madsen O, et al. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science, 2005,307: 580−584.
[2]Calisher CH, Childs JE, Field HE, et al. Bats:important reservoir hosts of emerging viruses. Clin Microbiol Rev, 2006, 19: 531−545.
[3]Freuling CM., Beer M, Conraths FJ, et al. Novel lyssavirus in Natterer's bat, Germany. Emerg Infect Dis, 2011, 17: 1519−1522.
[4]Kuzmin IV, Mayer AE, Niezgoda M, et al. Shimoni bat virus, a new representative of the Lyssavirus genus. Virus Res, 2010, 149: 197−210.
[5]Ge X, Li J, Peng C, et al. Genetic diversity of novel circular ssDNA viruses in bats in China. J Gen Virol, 2011, 92: 2646−2653.
[6]Du L, Lu Z, Fan Y, et al. Xi River virus, a new bat reovirus isolated in southern China. Arch Virol,2010, 155: 1295−1299.
[7]Chu DK, Poon LL, Guan Y, et al. Novel astroviruses in insectivorous bats. J Virol, 2008, 82:9107−9114.
[8]Marsh GA, de Jong C, Barr JA, et al. Cedar virus: a novel Henipavirus isolated from Australian bats.PLoS Pathog, 2012, 8: e1002836.
[9]Shi Z, Hu Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res,2008, 133: 74−87.
[10]Li W, Shi Z, Yu M, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science, 2005, 310:676−679.
[11]Augustina A, Heather J, Baldwin, et al. Human betacoronavirus 2c EMC/2012-related Viruses in Bats, Ghana and Europe. Emerg Infect Dis, 2013,19: 456−459.
[12]Zhang HL, Shi HF, Liu LH, et al. Isolation of chikungunya virus from bat in Yunnan province and serological investigations. Chin J Virol, 1989, 5(1):31−36 (in Chinese).
张海林, 施华芳, 刘丽华, 等. 从云南省蝙蝠中分离基孔肯亚病毒及血清抗体调查. 病毒学报,1989, 5(1): 31−36.
[13]Zhao CS, Jiang LH, Yu XL, et al. Isolation of Ross River Virus and its Antibody Prevalance in Hainan Province. Chin J Vet Sci, 1997, 17(3): 241−243 (in Chinese).
赵春生, 蒋廉华, 余兴龙, 等. 从海南省蝙蝠脑中分离出1株罗斯河病毒及其血清抗体调查. 中国兽医学报, 1997, 17(3): 421−423.
[14]Zhang HL, Zhang YZ, Huang WL, et al. Isolation of Japanese encephalitis virus from brain tissue of bat in Yunnan province. Virol Sin, 2001, 16(1):74−77 (in Chinese).
张海林, 张云智, 黄文丽, 等. 从云南省蝙蝠脑组织中分离出乙型脑炎病毒. 中国病毒学, 2001,16(1): 74−77.
[15]Li Y, Ge X, Zhang H, et al. Host range, prevalence,and genetic diversity of adenoviruses in bats. J Virol, 2010, 84: 3889−3897.
[16]Wang X, Qi D, Hu J. Recent researches on Chiroptera in China. Sichuan J Zool, 2004, 23(2):153−157 (in Chinese).
王晓琴, 齐敦武, 胡锦矗. 中国翼手目研究进展.四川动物, 2004 , 23(2): 153−157.
[17]Djikeng A, Kuzmickas R, Anderson NG, et al.Metagenomic analysis of RNA viruses in a fresh water lake. PLoS ONE, 2009, 4: e7264.
[18]Schoenfeld T, Patterson M, Richardson PM, et al.Assembly of viral metagenomes from yellowstone hot springs. Appl Environ Microbiol, 2008, 74:4164−4174.
[19]Tang P, Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol, 2010, 5:177−189.
[20]Lu J, Domingo JS. Turkey fecal microbial community structure and functional gene diversity revealed by 16S rRNA gene and metagenomic sequences. J Microbiol, 2008, 46: 469−477.
[21]Day JM, Ballard LL, Duke MV, et al. Metagenomic analysis of the turkey gut RNA virus community.Virol J, 2010, 7: 313.
[22]Donaldson EF, Haskew AN, Gates JE, et al.Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol, 2010, 84: 13004−13018.
[23]Li L, Victoria JG, Wang C, et al. Bat guano virome:predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol, 2010,84: 6955−6965.
[24]Ge X, Li Y, Yang X, et al. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol,2012, 86: 4620−4630.
[25]Wu Z, Ren X, Yang L, et al. Virome analysis for identification of novel Mammalian viruses in bat species from Chinese provinces. J Virol, 2012, 86:10999−11012.
[26]Tamura K, Peterson D, Peterson N, et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol,2011, 28: 2731−2739.
[27]Li Y, Ge X, Hon CC, et al. Prevalence and genetic diversity of adeno-associated viruses in bats from China. J Gen Virol, 2010, 91: 2601−2609.
[28]He B, Tu CC. The advances and applications of viral metagenomics. Acta Vet et Zoo Sin, 2012,43(12): 1865−1870 (in Chinese).
何彪, 涂长春. 病毒宏基因组学的研究现状及应用. 畜牧兽医学报, 2012, 43(12): 1865−1870.
[29]Cui J, Tachedjian M, Wang L, et al. Discovery of retroviral homologs in bats: implications for the origin of mammalian gammaretroviruses. J Virol,2012: 86: 4288−4293.
[30]International Committee on Taxonomy of Viruses.Master, Species, List 2011 v2. [EB/OL]. [2012-12-15]http://ictvonline.org/virusTaxonomy.asp?bhcp=1
[31]Allander T, Tammi MT, Eriksson M, et al. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci USA,2005, 102: 12891−12896.
[32]Campe H, Hartberger C, Sing A. Role of human bocavirus infections in outbreaks of gastroenteritis.J Clin Virol, 2008, 43: 340−342.
[33]Zhai S, Yue C, Wei Z, et al. High prevalence of a novel porcine bocavirus in weanling piglets with respiratory tract symptoms in China. Arch Virol,2010, 155: 1313−1317.
[34]Ganesh B, Banyai K, Martella V, et al.Picobirnavirus infections: viral persistence and zoonotic potential. Rev Med Virol, 2012, 22:245−256.
[35]Benko M, Harrach B. Molecular evolution of adenoviruses. Curr Top Microbiol Immunol, 2003,272: 3−35.
[36]McNeill KM, Ridgely Benton F, Monteith SC, et al. Epidemic spread of adenovirus type 4-associated acute respiratory disease between U.S. Army installations. Emerg Infect Dis, 2000, 6: 415−419.
[37]Jawetz E. The story of shipyard eye. Br Med J,1959, 1: 873−876.
December 19, 2012; Accepted: March 5, 2013
Changchun Tu. Tel/Fax: +86-431-86985862; E-mail: changchun_tu@hotmail.com NSFC云南省联合基金 (No. U1036601) 资助。
Metagenomic analysis of bat virome in several Chinese regions
Fanli Yang1,2, Yiyin Wang3, Wencheng Zheng4, Biao He2, Tinglei Jiang5, Yingying Li2,Lele Xia2, Ye Feng2, Quanshui Fan3, and Changchun Tu2
1College of Animal Science and Veterinary Medicine,Jilin University,Changchun130062,Jilin,China
2Institute of Military Veterinary,Academy of Military Medical Sciences,Changchun130122,Jilin,China
3Center for Disease Control and Prevention,Chengdu Military Region of PLA,Kunming650118,Yunnan,China
4The Animal Health Inspection of Hunan Province,Changsha410007,Hu’nan,China
5Jilin Key Laboratory of Animal Resource Conservation and Utilization,Northeast Normal University,Changchun130024,Jilin,China
viral metagenomic, bat, virome, next-generation sequencing
Supported by: NSFC-Yunnan Province Joint Fund (No. U1036601).
时间:2013-04-10 网络出版地址:http://www.cnki.net/kcms/detail/11.1998.Q.20130410.1005.003.html
Bats are important reservoir animals and more than 60 viruses have been identified in bats with many of them highly pathogenic to human. In order to understand the natural background, genetic diversity of bat viruses in China and discover potential viral pathogens, Solexa sequencing based viral metagenomics focusing on bats tissues was established and to analyze the virome of bats collected from Jilin, Yunnan and Hunan province. By Solexa sequencing, 116 442 324 useful reads were obtained and assembled into 4 872 contigs, of which 8.2% (4 002/4 4872) were annotated to 36 viral families, including 19 vertebrate virus families, 6 plant virus families, 4 insect virus families and 4 phages. Further contigs analyses showed that some adenovirus, bocavirus, picobirnavirus, parvovirus contigs sequences were similar with known viruses. However, part of them shared limited identities to these viruses implying the discovery of new viruses. Moreover,PCR validation of adenovirus and bocavirus confirmed the results obtained by viral metagenomics. This study aimed to understand bat virome in China by viral metagenomics and could be helpful to establish effective surveillance on wildlife-associate zoonoses.
(本文责编 郝丽芳)
