去氢芳樟醇合成去氢乙酸芳樟酯的工艺优化
2013-07-19李莹
李 莹
( 中国石化上海石油化工股份有限公司精细化工部,200540)
去氢乙酸芳樟酯是一种重要的中间体,可用于生产柠檬酸、紫罗兰酮等精细化工产品[1]。对去氢乙酸芳樟酯进行加氢即可得到乙酸芳樟酯。乙酸芳樟酯香气近似香柠檬油和薰衣草油,芬芳而幽雅,在各种花香香料的调香中有广泛的用途[2]。
去氢芳樟醇是含三键的不饱和脂肪叔醇,比含有乙烯基叔醇结构的芳樟醇稳定。同时,去氢芳樟醇酯化反应的逆反应速率常数相对比较低,有利于提高酯化反应的转化率和选择性,最终提高乙酸芳樟酯的收率[1]。因此,将去氢芳樟醇酯化、加氢是合成乙酸芳樟酯的一种更为理想的工艺路线[3-4]。该工艺路线中,由去氢芳樟醇酯化制备去氢乙酸芳樟酯是合成乙酸芳樟酯的关键。文章以去氢芳樟醇为原料,对通过酯化反应合成去氢乙酸芳樟酯的工艺条件进行研究。
1 实验部分
1.1 实验原料
实验所用原料均为分析纯,具体见表1。
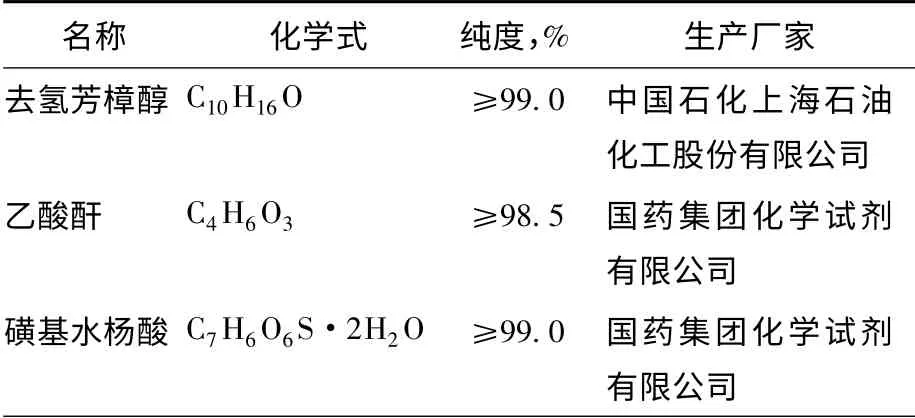
表1 实验原料一览表

续表1
1.2 反应原理
去氢芳樟醇与乙酸酐在催化剂的作用下进行酯化反应制备去氢乙酸芳樟酯,主要反应式如下:
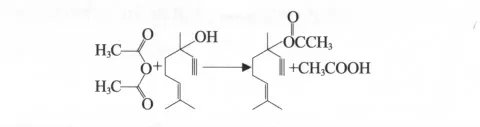
去氢芳樟醇与乙酸酐反应除了生成目的产物去氢乙酸芳樟酯及副产物乙酸外,酯化过程中也伴随异构、环化等副反应,生成环状吡喃和环状酯等副产物。
1.3 实验步骤
对原料进行称重,将部分原料按一定配比加入带有搅拌、温度计、冷凝器和恒压滴液漏斗的四口烧瓶,开启搅拌器,设定好恒温水浴槽温度,开始加热。待温度上升到设定温度后,加入所需催化剂,并缓慢滴加余下的原料,控制滴加速度,尽量使反应温度平稳上升。滴加完毕后继续反应至结束,停止加热,冷却至室温后停止搅拌,对产物进行称重及后续处理。反应过程中定时取样,样品用稀碱液和水洗涤至中性后,用气相色谱仪分析反应液组成。
1.4 产品组成分析方法
采用Agilent 6890 型气相色谱仪分析产品的组成,色谱柱型号为HP -1,规格为50 m ×200 μm×0.50 μm。用安捷伦化学工作站进行数据处理,按校正面积归一化法定量分析各组分的质量浓度,计算反应物的转化率及目的产物的选择性。
气相色谱分析操作:柱温在120 ℃下保持5 min,然后以15 ℃/min 升温速率升温至165 ℃,保持8 min;继续以20 ℃/min 的升温速率升温至220 ℃,保持15 min。采用氢火焰离子化检测器,检测器温度为240 ℃,进样温度240 ℃,分流比100∶1,进样量0.4 μL,柱流量0.5 mL/min。
1.5 数据处理方法
去氢芳樟醇转化率y 按下式进行计算:
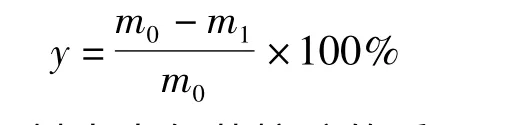
式中:m0为原料中去氢芳樟醇的质量g;m1为产物中去氢芳樟醇的质量,g。
2 结果与讨论
2.1 去氢芳樟醇酯化反应催化剂的选择
在化学反应中,催化剂之所以能显著地加快反应速率,是因为它改变了反应的历程,使反应的活化能显著降低,增加了活化分子百分数,从而使反应速率大大增加。去氢芳樟醇酯化反应常用的催化剂有对甲苯磺酸、磺基水杨酸、酸性阳离子交换树脂及杂多酸催化剂等。不同催化剂的反应结果见表2。
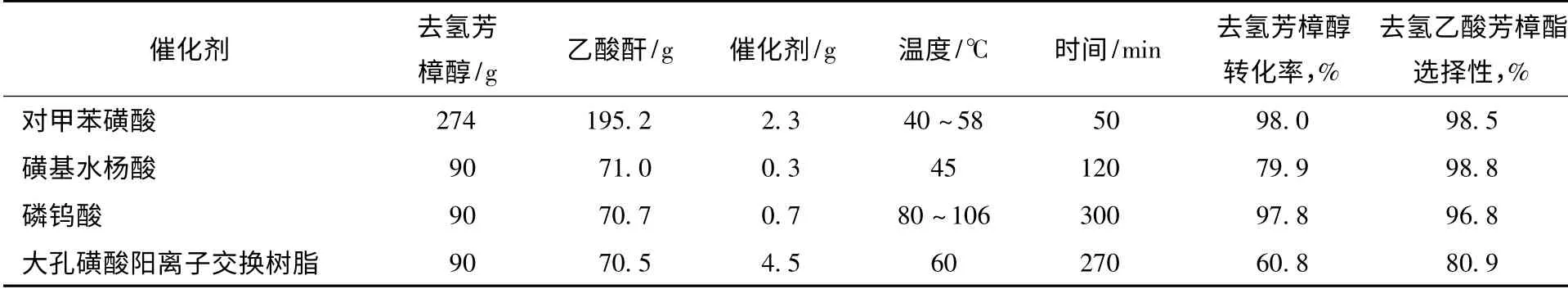
表2 不同催化剂对酯化反应的影响
从表2 可以看出:
(1)以大孔磺酸阳离子交换树脂为催化剂时,由于乙酸酐与树脂存在一定反应,乙酸酐消耗量大,尽管在相对较高的温度下反应,去氢芳樟醇转化率仍然较低(仅为60%),副反应较多,反应选择性也不高(仅为80%),反应效果较差。
(2)以磷钨酸为催化剂时,需要在较高温度下进行长时间反应,反应转化率及目的产物的选择性才能达到要求。因此,用磷钨酸作催化剂时能耗相对较高。
(3)以磺基水杨酸为催化剂,在实验条件下反应生成去氢乙酸芳樟酯的选择性较好,但转化率不高,仅为80%左右,催化剂的活性较低,反应效果一般。
(4)以对甲苯磺酸为催化剂时,在实验条件下催化剂表现出较高的反应活性及良好的去氢乙酸芳樟酯选择性。
因此,合成工艺条件的优化研究主要以对甲苯磺酸为催化剂。
2.2 原料配比对反应的影响
在其他条件不变的情况下,反应物物质的量浓度越大,单位体积内活化分子数增多,有效碰撞次数增加,反应速率越大。因此,适当提高原料配比中乙酸酐的比例可提高去氢芳樟醇的转化率。
在催化剂加入量为去氢芳樟醇质量的0.6%、反应温度40 ℃的条件下,缓慢滴加去氢芳樟醇,滴加完毕后继续保持恒温反应,反应时间100 min,考察不同醇酐物质的量比对酯化反应的影响,结果见表3。

表3 原料配比对酯化反应的影响
增加乙酸酐的比例可以提高去氢芳樟醇的转化率,同时副反应随着转化率的提高而增加,目标产物的选择性降低。此外,乙酸酐配比过大,会增加未反应的乙酸酐的处理量。综合以上结果,酯化反应适宜的醇酐的物质的量比为1∶1.1 ~1∶1.2。
2.3 催化剂加入量对反应的影响
催化剂的加入量直接影响反应速度和反应产物的选择性。在反应温度40 ℃、去氢芳樟醇与乙酸酐的物质的量比为1∶1.2、反应时间2 h 的条件下,实验结果如图1 所示。
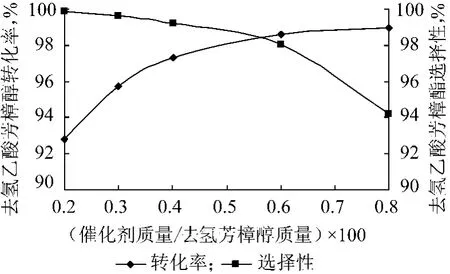
图1 催化剂加入量对酯化反应的影响
从图1 可以看出:去氢芳樟醇的转化率随着催化剂用量的增加而增加,当催化剂加入量为去氢芳樟醇的0.6%后,上升趋势变缓;生成去氢乙酸芳樟酯的选择性则与之相反,随着催化剂加入量的增加而下降。这是因为催化剂可以降低反应活化能,不仅加快主反应速度,副反应速度也随之加快。因此,催化剂比例高,反应速度快,去氢芳樟醇转化率增加,但去氢乙酸芳樟酯的选择性随之下降。因此,催化剂加入量为去氢芳樟醇质量的0.5% ~0.6%比较适宜。
2.4 反应温度对反应的影响
反应温度是影响化学反应速率的重要因素之一,温度升高往往会加速反应的进行。这是因为温度的升高使一些能量较低的分子获得了能量,从而成为活化分子,增加了反应物中活化分子数,大大提高了反应的速度。
在催化剂加入量为去氢芳樟醇质量的0.6%、去氢芳樟醇与乙酸酐的物质的量比为1∶1.2、反应时间2 h 的条件下,实验结果如图2 所示。
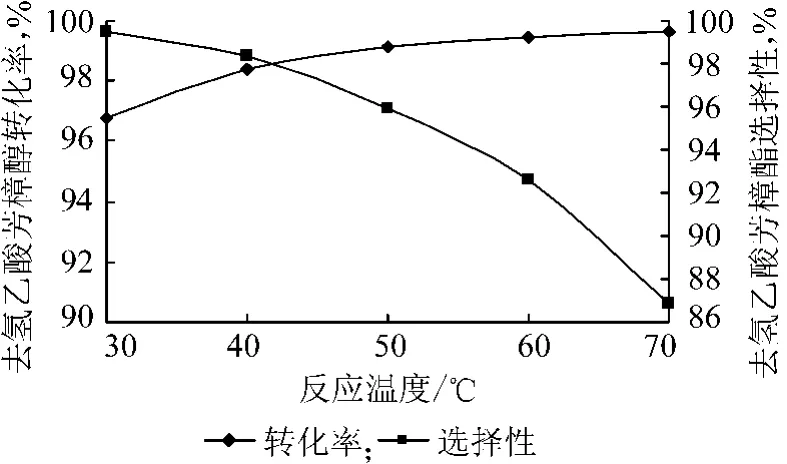
图2 反应温度对酯化反应影响
从图2 中可以看出:随着反应温度的升高,去氢芳樟醇的转化率增加,反应的选择性则随反应温度的上升而呈单调下降趋势。这是因为去氢芳樟醇与乙酸酐进行酯化反应,是放热反应,温度较低有利于主反应的进行,反应的选择性更高;而反应温度的上升更利于副反应的进行,从而使反应的选择性下降。
酯化反应选择在较低的温度下进行可以提高目标产物的选择性,但反应速度变慢,需要较长时间来提高去氢芳樟醇的转化率。实验结果表明在35 ~40 ℃下反应的转化率和去氢乙酸芳樟酯的选择性均较高(98%左右)。
2.5 反应时间的影响
在反应温度40 ℃去氢芳樟醇与乙酸酐的物质的量比为1∶1.2、催化剂加入量为去氢芳樟醇质量的0.6%的条件下,考察反应时间对酯化反应的影响,实验结果如图3 所示。
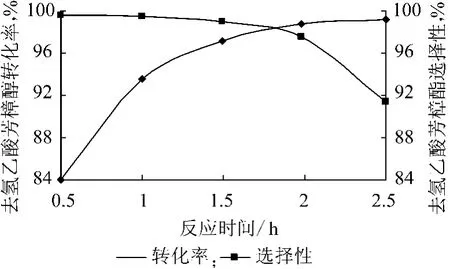
图3 反应时间对酯化反应的影响
从图3 可以看出:去氢芳樟醇的转化率随反应时间的增加快速上升,到反应后期上升速度变缓;去氢乙酸芳樟酯的选择性则随着反应时间的增加而缓慢下降,反应后期选择性下降幅度较大。这是因为在反应体系中大部分去氢芳樟醇转化为酯后,随着反应时间的增加,酯的异构体比例及重组分开始上升,反应的选择性下降较快。反应时间为2 h 时转化率和选择性均大于98%。
上述研究结果表明:在去氢芳樟醇与乙酸酐的物质的量比为1∶1.1 ~1∶1.2、催化剂加入量为0.5% ~0.6%、反应温度35 ~40 ℃、反应时间为2 h 的条件下,反应转化率及去氢乙酸芳樟酯的选择性均不小于98%。
3 副产物乙酸的回收
从酯化反应的原理中可以看出:酯化反应过程中同时有副产物乙酸生成。在已报导的文献中,乙酸通常不回收,而是直接加以水洗,再用一定浓度的Na2CO3溶液进行碱洗,将其完全脱除。由于这种处理方法废水的处理量很大,在工业化生产上是不可取的。因此,必须考虑乙酸的回收问题。
若要对乙酸进行回收,一般的方法是在含有乙酸的水相中加入共沸剂,通过共沸精馏将水脱除,最后可得到高纯度的冰乙酸,但此方法能耗大,因此也是不可取的。
本实验将酯化反应产物用旋转蒸发仪蒸馏而快速分离,得到富含乙酸的馏分,再经过精馏,得到高纯度的乙酸。富含去氢乙酸芳樟酯的釜液用10% 的Na2CO3溶液洗涤至中性,经过减压精馏,得到纯度98%以上的去氢乙酸芳樟酯产品,用作选择性加氢的原料。
该法操作简单,很容易得到质量分数大于99.8%的可直接作为产品销售的高纯度乙酸。操作在减压条件下进行,在绝对压力为1.0 kPa、回流比为2∶2、顶温15 ℃及釜温45 ℃左右时,接收该馏分即可得到上述乙酸产品,乙酸收率大于95%。
该法操作条件温和,釜液中的去氢乙酸芳樟酯通过集中精馏实现回收,提高了酯化产物的总收率,且废水处理量少,具有较高的工业化应用价值。
4 结论
(1)选择对甲苯磺酸作为去氢芳樟醇与乙酸酐酯化反应的催化剂。该催化剂具有反应活性高、用量省及酯化反应温度低等优点。
(2)对酯化反应工艺进行优化,确定醇酐的物质的量比为1 ∶1. 1 ~1 ∶1. 2,催化剂加入量为去氢芳樟醇质量的0. 5% ~0. 6%,反应温度为35 ~40 ℃,反应时间为2 h。在此条件下,去氢芳樟醇的转化率和去氢乙酸芳樟酯选择性均超过98%。
(3)对副产物乙酸的回收工艺进行了研究,打通了以去氢芳樟醇与乙酸酐为原料合成去氢乙酸芳樟酯的全流程,为工业化应用提供技术参考。
[1] 王磊君,朱志庆,吕自红.强酸性阳离子交换树脂催化合成乙酸脱氢芳樟酯[J]. 化工生产与技术,2010,17(5):31 -33.
[2] 刘亚涛,朱志庆,吕自红.由芳樟醇合成乙酸芳樟酯的研究进展[J].化学世界,2003,44(6):327 -331.
[3] Birbiglia James A,Chase George O,Julius Galende. Teritary esters and preparation thereof:US,2797235[P].1954-09-01.
[4] Faleeva L,Sidorov I,Keller G. Preparation of linalyl acetate from dahydrolinalyi acetate[J]. Maslo - Zhir Prom - st,1975(7):32 -34.
