塞来昔布的合成、药理作用和临床应用研究进展
2013-07-02姚世宁徐畅张青青黄华姚其正
姚世宁徐 畅张青青黄 华姚其正*
(1 江苏伊斯特威尔国际贸易有限公司,市场研发部,江苏 南京 210002;2 中国药科大学药学院,江苏 南京 210009)
塞来昔布的合成、药理作用和临床应用研究进展
姚世宁1徐 畅2张青青2黄 华1姚其正2*
(1 江苏伊斯特威尔国际贸易有限公司,市场研发部,江苏 南京 210002;2 中国药科大学药学院,江苏 南京 210009)
综述了近10多年来具有代表性的COX-2抑制剂塞来昔布合成方法,并对合成方法的优缺点进行了剖析,同时阐述了制备塞来昔布过程中常形成的晶型与杂质;对于塞来昔布的药理学作用、临床疗效和副作用作了归纳总结。
塞来昔布;COX-2抑制剂;合成;晶型;杂质;药理学;临床作用;副作用;综述
1 概 述
塞来昔布(Celecoxib,1)是1,5-二芳基取代吡唑类化合物,化学名为4-[5-(4-甲基苯基)-3-(三氟甲基)-1H-吡唑-1-基]苯磺酰胺,pKa值为11.1。塞来昔布商品名为“西乐葆”(Celebrex),是由Pharmacia公司开发的,于1998年12月31日通过FDA审批上市的第一个选择性环氧合酶-2(COX-2)抑制剂,1999年正式应用于临床。2003年Pfizer通过收购Pharmacia获得塞来昔布的胶囊制剂西乐葆。
塞来昔布可用于风湿性关节炎、骨关节炎及这些疾病引起的疼痛治疗,效果与其他非甾体抗炎药(NSAIDs)如萘普生、布洛芬相当,但是相对于传统非选择性的非甾体抗炎药(如萘普生、布洛芬),塞来昔布引起上消化道并发症的几率大大降低[1],根据IMS Health Inc.的报道和Pfizer公司的年报,2009年辉瑞Celebrex全球销售额23.8亿美元,保持全球抗风湿病处方药第一的品牌地位。2006~2012上半年,Celebrex全球年销售额稳定,为24亿美元左右;2012上半年,Celebrex全球销售额相对去年同比上升了6%。在欧美等国外市场上,Celebrex在抗风湿类药物中占有率稳定,为23%左右。2011年我国塞来昔布的市场份额为8%,位列市场用药的第4位。
2 塞来昔布的合成
2.1 塞来昔布逆合成分析与合成路线
塞来昔布(1)是1,5-二芳基取代吡唑类化合物,其逆合成分析的切断位置计有5处(图1),主要形成以下3种切断组合和相应的合成片断:
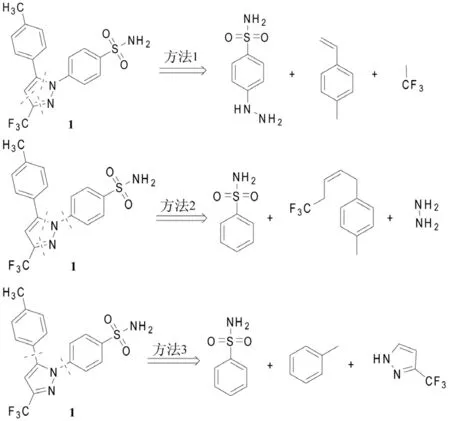
图1 塞来昔布(1)的逆合成分析
根据这几种不同的切断组合,现对近10多年来具有代表性的塞来昔布合成方法,以及各合成方法的优缺点进行总结和剖析。
2.2 芳基取代肼的环合反应(方法1)
方法1:主要是以对磺酰胺基芳肼为原料,与不同切断后的合成等价试剂经缩合与环合,制得目标吡唑类衍生物——塞来昔布(1),由此有以下多步合成化合物1的三条路线。
2.2.1 路线一(Scheme 1)[2,3]
Penning等[2]用甲苯酮2与三氟乙酸乙酯(3)在25% NaOMe/MeOH和甲基叔丁醚的混合液中进行Claisen缩合,得到收率为94%的苯基取代的1,3-二羰基化合物4,然后与对磺酰胺基苯肼(5)在HCl/EtOH中回流;反应结束后,经快速柱层析分离去除区域异构体20(其结构见Scheme 4),得到塞来昔布(1),总收率为43%。也可用庚烷对粗产品1进行重结晶去除了异构体20,不用柱层析纯化,适合大规模生产,这为商业化生产塞来昔布提供了方法。
Reddy等[3]用乙酸乙酯和水为混合溶剂,将化合物4与5的盐酸盐缩合-环合,经甲苯重结晶后,塞来昔布(1)单步产率可达84%,此方法产生较少的区域异构体20副产物,纯化后的塞来昔布固体中所含异构体20少于0.03%。
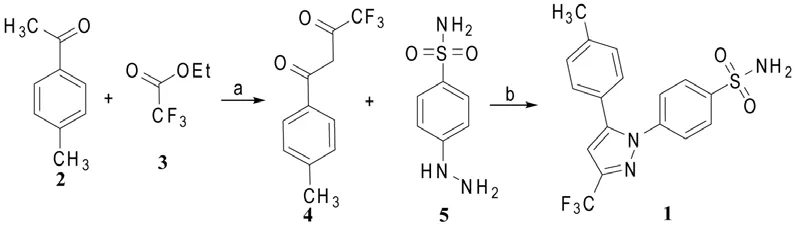
Scheme 1 试剂和条件[2]:a:①NaOMe/MeOH, rt;②回流;b:HCl/ EtOH:回流
2.2.2 路线二(Scheme 2)
Reddy等[4]将甲苯(7)与三氟甲基取代的α,β-烯酮8的二氯甲烷溶液、氯化锌混合,22℃下反应3 h,制得两端取代的α,β-烯酮9,将9与溴在室温下反应30min,后在KOH-乙醇溶液中回流3h,得产品α,β-炔酮10与对磺酰胺基苯肼(5)盐酸盐在乙醇中回流反应4 h,经重结晶,制得塞来昔布(1)。此路线原料价廉易得,产率高,具有较好的区域选择性[化合物1∶化合物20(w/w)=9∶1]。
2.2.3 路线三(Scheme 3)
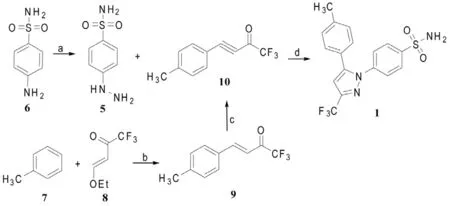
Scheme 2 试剂和条件[4]:a:①HCl,NaNO2/H2O,0°C;②HCl,SnCl2/ H2O,冷却;b:ZnCl2/CH2Cl2,22°C,3h;c:①Br2/CHCl3,rt,②KOH/ EtOH,回流,3h;d: EtOH,回流
在上述两法环合时都存在着区域选择性问题,为了避免产生区域异构体20,Oh[5]采用1,3-偶极环合法合成塞来昔布(1)。首先以对磺酰胺基苯肼盐酸盐(11)与三氟乙酸酐(12)为原料,在乙腈中5-10℃反应4 h,得到的三氟乙酰苯肼13在5-10℃下和苯磺酰氯(14)反应1 h,制得1,3-偶极化合物——苯磺酸酯衍生物15,从而确定了CF3的位置,为不产生区域异构体20打下基础。
化合物15与事先制备的乙烯衍生物17混合,反应10min,环合,得粗产品1,用异丙醇-水重结晶,得高纯度的塞来昔布(1),总产率为52%。
此路线具有高度的区域选择性,无区域异构体20产生,该方法合理、简单与实用,所用原料和试剂价较廉易得。

Scheme 3 试剂和条件[5]:a:MeCN,5~10℃,4h;b:AcOEt/N-甲基吗啡啉,5~10℃; c: ①TiCl4,Na2SO4/PhMe,5℃,②EtN(Pr-i)2,rt→70℃→5℃;d:①Et3N/THF,5~10℃,②HCl/H2O,AcOEt
2.3 “一锅煮”法制备塞来昔布(方法2)
路线四(Scheme 4):
Beveridge等[6]用5 mol % Cu(OAc)2H2O作催化剂,使偶氮二甲酸叔丁酯(18)与硼酸衍生物19偶联,制得的苯肼类衍生物不分离,直接与1,3-二羰基化合物4环化,“一锅煮”法获得塞来昔布(1)与其区域异构体20(2:1)的混合物,产率为35%(以混合物计)。
此法相对于“2.2”节中的取代苯肼与1,3-二羰基化合物环合制备吡唑类化合物的方法,操作简便,可以认为是一步反应,苯肼类衍生物边产生边被环合;而且“2.2”节环合法中所需取代苯肼衍生物的制备也较困难。此路线所需苯硼酸类衍生物可通过商业途径获得,硼酸类前体即使合成,其方法亦较温和。
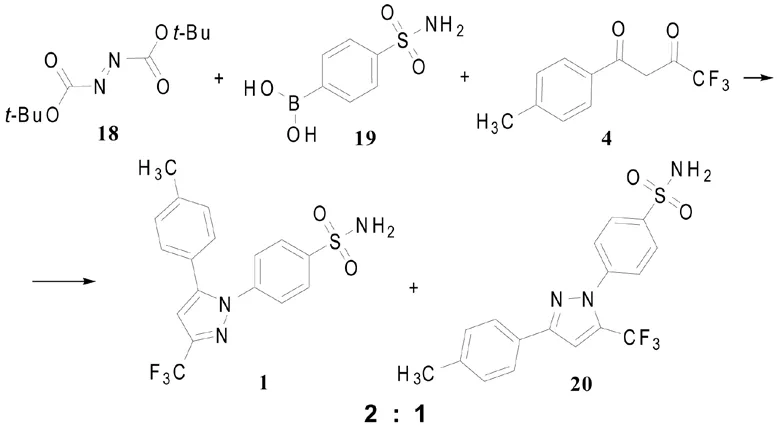
Scheme 4 试剂和条件[9]:1)Cu(OAc)2/MeOH, 65℃,65℃→rt,2)HCl/二氧六环,rt,80℃
2.4 吡唑环的直接芳基化反应(方法3)
路线五(Scheme 5):
Gaulier等[7]以3-三氟甲基-1H-吡唑(24)为原料,在1,4-二氧六环中,与对碘苯磺酰氯(21)和二苯甲基胺(22)制得的中间体23反应,得取代吡唑中间体25,再以醋酸钯为催化剂,与对溴甲苯(26)偶联,经柱层析色谱纯化后,无区域异构体。用浓硫酸将27脱保护,制得塞来昔布(1),总产率33%,产品纯度95%以上。此合成路线采用3-三氟甲基-1H-吡唑的直接芳基化的方法,提高了区域选择性,抑制了区域异构体含量。

Scheme 5 试剂和条件[7]: a:Et3N/CH2Cl2,rt,b:1)K3PO4/二氧六环,rt→80℃,2)C6H11NMe2,CuI,80℃,115℃,c: K2CO3,正丁基-二(1-金刚烷基)膦,t-BuCO2H,Pd(OAc)2/AcNMe2,140℃, d: H2SO4,rt.
2.5 合成路线总结
在以上合成塞来昔布(1)的路线中,路线一是较为成熟的路线,对其反应条件改进的文献也较多,包括溶剂选择、反应温度的优化、区域异构体的减少与分离等方面,适合工业化生产。路线二与路线一原理基本相同,但是使用了NaNO2、Br2等试剂,会造成较大的污染,且相对路线一没有明显改进。路线三的区域选择性得到了显著提高,但是反应过程需要反复保护与脱保护,较为繁琐,第一步以乙腈为溶剂,污染较大。路线四明显的缺陷是合成中产生了大量区域异构体20,不过此合成路线较短,“一锅煮”法,操作简便。路线五具有全新的思路,并且可达到无区域异构体20产生,但是塞来昔布(1)的总产率较低,所用催化剂如正丁基-二(1-金刚烷基)膦、Pd(OAc)2成本较高,脱保护基要用浓硫酸,后处理麻烦,纯化方式主要为柱层析法,仅适合实验室小规模制备。
除路线一以外,路线三、路线五有一定的优势,这2条合成路线致力于从根本上扼制区域异构体20的产生,但成本较高,步骤较多,总产率不高,要使它们成为工业化生产的主流方法,需要花大力气优化反应条件。综上所述,现在芳基取代肼环化反应的路线一仍为生产塞来昔布不可替代的工业化路线,但仍有进一步工艺改进优化的余地。
3 晶型与杂质
3.1 晶型
塞来昔布常见结晶形态有3种。一般生产过程所得的塞来昔布为热力学稳定的晶型3[8],以三斜晶空间群描述的晶体学参数为a=10.136(5)Å,b=16.778(6)Å,c=5.066(6)Å,α=97.62(7)º,β=100.65(6)º,γ=95.95(4)º[9]。塞来昔布溶解于甲醇,活性炭脱色,过滤,向滤液中加入适量水,25~30℃放置3~5h,过滤、以水洗涤、干燥,得塞来昔布晶型3,X射线粉末衍射图显示在5.4、9.0、9.8、10.7、11.0、13.0、14.9、16.1、18.0、19.7、21.5、22.2和22.5º2θ处有峰[10]。晶型3为长针状结晶晶态,在压片时塞来昔布易熔化后凝结成块,与辅料分离,产生不均匀的混合物。
晶型1的X射线粉末衍射图显示在5.5、5.7、7.2和16.6º2θ处有峰,熔点为162.8℃,其红外光谱在3250cm-1至3260cm-1及3350cm-1至3360cm-1处有峰。
晶型2的X射线粉末衍射图显示在10.3、13.8或17.7º2θ处有峰,熔点大约在161.5℃。晶型1和晶型2相对晶型3具有更高的溶解度与更快的溶解速度,这是因为晶型3较晶型1和晶型2热力学稳定,具有更低的自由能。溶解速度的加快可以提高药物的生物利用度。晶型3 的熔点为160.8℃。在熔化过程中,晶型3可以部分转化为晶型1或晶型2[8,11]。
文献8提供的结晶方法难以高纯度、可重复地、大规模地生产单一晶型的塞来昔布,所得产品含有无定型的以及热力学稳定的晶型3塞来昔布,这种产品不稳定,放置中含晶型3塞来昔布会逐步增加。而将塞来昔布晶型3悬浮于加热的正癸烷后冷却,加入晶种后继续冷却,过滤并干燥可得晶型1的塞来昔布,将固有溶解速率从晶型3的8μg/min提高到晶型1的11μg/min。
另外,还有晶型4和晶型5。将塞来昔布溶解在酰胺类溶剂如N,N-二甲基甲酰胺(DMF)中,再引入反溶剂如水,可得到晶型4。X射线粉末衍射图显示在11.0、18.3、23.2、27.6、30.0、35.0和37.9º2θ处有峰。晶型4比晶型3具有更高的堆积密度与振实密度(见表1),在制备小剂量制剂时有优势[12]。见表1。

表1 晶型3和4的堆积密度、振实密度对比17
N,N-二甲基乙酰胺(DMA)为溶剂诱导结晶,可得塞来昔布晶型5,其红外光谱在3997、3128、1604、1471、1404、1342、1269、1233、1127、1098、972、838、816和597cm-1处有吸收,X射线粉末衍射图显示在8.60、12.9、15.7、18.0、19.7、21.1、22.5、23.1、24.7、23.1、24.7、25.9、27.3、29.5和30.1,º2θ处有峰,熔点145-149℃。还可以二甲基亚砜(DMSO)、1-甲基吡咯烷酮(NMP)为溶剂诱导结晶[13]。
3.2 杂质[14-16]
见图2。商业生产路线所得的塞来昔布含有杂质化合物2、4、5、20、4-甲基苯甲酸甲酯(28)、5-(4-甲基苯基)-3-三氟甲基-1H-吡唑(29)和4-[5-(2-甲基苯基)-3-(三氟甲基)-1H-吡唑-1-基]苯磺酰胺(30)七种(Fig. 2)。根据HPLC分析[15],2、5、28和30的极性大于塞来昔布,而20相对塞来昔布为非极性的;塞来昔布原料药粗品含杂质量常>2%,其中,2所占含量百分比可忽略不计,28为0.05%,29为0.15%,30为0.1%,20为0.38%,可见20为主要杂质。

图2 塞来昔布中的杂质
杂质来源和形成分析:2是塞来昔布反应的起始原料;28是起始原料2中所含杂质;4和5是合成塞来昔布的中间体。在最后一步缩合反应中,中间体5中所含的单质肼盐与4缩合——这个平行反应可能是产生29的原因。20和30均产生于合成塞来昔布的最后一步,即4和5缩合时形成的异构体。20为区域异构体,30成为邻位异构体(由4中含有的邻位异构体杂质与5环合形成的);而2、4、5、28和29都是前期生产过程带入的,体现严格控制原料质量的重要性。
主要杂质的药理学与毒理学性质未知。一项基于COX-2酶结构的对接研究表明,在塞来昔布、20、5、4和2中,与COX-2酶结合度的排序为塞来昔布>20>5>4>2,可见区域异构体结合度良好,若这样的杂质在原料药中超量布,它与酶结合后所造成的影响难以忽视和估量,可见合成中对杂质的控制与产品的纯化至关重要。但目前市售塞来昔布胶囊中的杂质含量并不显著[16]。
4 药理作用与临床应用
4.1 药理作用[17-19]
塞来昔布对环氧合酶(COX)-2的选择性因分析方法不同而改变。一项人体内多种重组酶分析结果显示,塞来昔布对COX-2的选择性是COX-1的375倍。然而,另一项人体内全血分析结果表明,塞来昔布对COX-2的选择性是COX-1的30倍,而布洛芬为0.5倍,萘普生0.7倍,吲哚美辛1.9倍,美洛昔康18倍,尼舒利美19倍,双氯高灭酸29倍,罗非昔布267倍。健康受试者服用塞来昔布治疗推荐剂量(200mg或400mg/d)后,凝血氧烷B2水平(COX-1活性指标)未见显著抑制,而体外脂多糖调节的前列腺素(PG)E2生成(COX-2活性指标)受到显著抑制。塞来昔布对于COX-2的选择性低于其他昔布类药物,比如罗非昔布、伐地考昔、罗美昔布。
在动物炎症实验模型中,塞来昔布表现为具有消炎镇痛活性,与其他非选择性非甾体抗炎药(NSAIDs,如吲哚美辛、萘普生、吡罗昔康)活性相似。在动物模型中,超治疗剂量的塞来昔布对消化道粘膜没有损伤;受试者每天2次、每次100mg或200mg、连续7天服用塞来昔布,所造成的胃部/十二指肠的腐蚀/溃疡显著少于萘普生,与安慰剂相似。
由于根本性地抑制血小板中的COX-1,NSAIDs可能会抑制凝血氧烷A2的生成,并损伤血小板凝集,加强了出血倾向。然而,几乎完全(95%)抑制血小板COX-1活性才会造成血小板凝集的抑制。超治疗剂量服用塞来昔布(800mg/d和1200mg/d)也只会部分抑制血小板COX-1,因此不会影响血小板凝集。
对血小板凝集的深度抑制是低剂量阿司匹林心脏保护作用的基础。与部分NSAIDs(如萘普生和布洛芬)不同,塞来昔布不会阻碍COX-1的活性位点,不会干扰低剂量阿司匹林的心脏保护作用。
非选择性COX抑制引起的PGs抑制会造成预先患有高血压的病人血压上升,尤其是这些病人服用的药物为ACE抑制剂和β-肾上腺素受体拮抗剂,因为这类抗高血压药部分作用机理为增加扩血管PGs的合成量。研究表明,与罗非昔布不同,塞来昔布不会引起治疗中高血压病人24h收缩压的显著增加,但是在少部分病人中会造成一定水平的血压控制不平稳。塞来昔布不会显著降低环前列腺素的生成。一个19项实验的荟萃分析显示,服用罗非昔布会比服用安慰剂或NSAIDs显著增加患高血压的风险,而塞来昔布造成的风险较安慰剂或NSAIDs低,但不显著。
COX-2在某些肾组织中持续表达,塞来昔布和NSAIDs都会抑制PGE2和6-氧化-PGF1α的分泌,并且都会对肾功能产生不良影响(如液体潴留和浮肿)。
4.2 临床应用
4.2.1 骨关节炎与风湿性关节炎
数项6~13周、安慰剂对照的动态校验实验表明,200mg/d或400mg/d剂量的塞来昔布(相对安慰剂)对髋部和/或膝部骨关节炎患者的症状治疗有显著效果。2~12周髋部或膝部骨关节炎症状治疗实验中,口服标准剂量(200mg/d或400mg/d)通常与一日口服3次50mg双氯芬酸,口服2次140mg双氯芬酸-考来烯胺,口服2次400mg右布洛芬或口服2次500mg萘普生同样有效。对于长达13或26周的髋部或膝部骨关节炎症状治疗,服用200mg/d的塞来昔布与服用其他昔布类,即30mg/d的艾托考昔和100~400mg/d的罗美昔布同等有效。24周、200mg/d服用塞来昔布对膝部症状性比安慰剂显著有效,而膳食补充氨基葡萄糖和硫酸软骨素与安慰剂对比无显著效果[17]。
在一系列6~24周随机控制实验中,200mg/d和400mg/d的塞来昔布与安慰剂对比[20],对风湿性关节炎患者有显著疗效,但与萘普生[20]、双氯芬酸[21,22]、美洛昔康[22],萘丁美酮[22]疗效无显著区别。
4.2.3 强直性脊椎炎
6或12周服用200mg/d或400mg/d塞来昔布可相对安慰剂显著缓解活动性强直性脊椎炎患者的疼痛,减轻功能性损害进一步发展,降低疾病活动性。总体而言,与非选择性NSAIDs相比,塞来昔布对治疗活动性强直性脊椎炎同等有效,例如400mg/d的塞来昔布与1000mg/d的萘普生疗效无显著区别[17]。
4.2.4 肿瘤
随机塞来昔布预防腺瘤(APC)实验分析了2035位高风险结肠腺瘤息肉患者,患者每日服用200mg2次(低剂量)或每日400mg 2次(高剂量)或安慰剂,安慰剂组三年腺瘤风险>60%,风险极高;低剂量塞来昔布组相对安慰剂组风险为33%,高剂量组为45%,风险显著下降;塞来昔布还可以减小腺瘤体积、较少数量、降低患者负担。第1年腺瘤风险降低就十分显著,至第3年降低程度仍然相似[23,24]。
4.3 副作用与耐受性
4.3.1 消化道副作用
在一项随机、控制变量的疗效实验中,骨关节炎、风湿性关节炎或强直性脊椎炎患者未见明显副作用,胃消化道副作用发生率以及因消化道副作用引起的停药与安慰剂、扑热息痛和NSAIDs相似。一些长达12~13周、使用内窥镜的实验表明,使用200mg/d或400mg/d塞来昔布治疗骨关节炎或风湿性关节炎而发生胃十二指肠溃疡的发生率显著低于使用2400mg/d的布洛芬,或使用1000mg/d的萘普生。12周服用200mg/d或400mg/d的塞来昔布的骨关节炎患者溃疡或上消化道综合症发生率也显著低于100mg/d双氯芬酸[22]。
4.3.2 心血管及肾脏副作用
APC实验显示,400mg/d和800mg/d的塞来昔布与安慰剂相比,呈剂量依赖性地增加了心血管风险,即患非致命性心肌梗塞(MI)、中风或心衰的风险。然而,结直肠散发性腺瘤息肉(PreSAP)腺癌预防实验却未显示400mg/d与安慰剂相比有显著增加如上心血管风险的效果。两个塞来昔布用于治疗关节炎主要病症的实验显示,200mg/d,400mg/d或800mg/d的塞来昔布相比安慰剂或非选择性NSAIDs(萘普生,布洛芬,双氯芬酸,洛索洛芬或酮洛芬)不会增加心血管风险。
北美一项长达12周的肾脏副作用混合分析显示[25],塞来昔布引起肾脏副作用的几率高于安慰剂,但与NSAIDs无显著区别。塞来昔布会引发肾脏副作用通常包括腺瘤、高血压和高血压的加剧,每个实验组有0.2-0.3%的患者因肾脏副作用撤出实验。
4.3.3 肝脏副作用
<200~800mg/d的塞来昔布造成的肝胆副作用发生率与安慰剂和2400mg/d布洛芬无显著区别,并且显著高于1000mg/d的萘普生。200mg/d塞来昔布肝脏副作用发生率为0.02%,400mg/d塞来昔布为0.04%,双氯芬酸为0.14%,布洛芬为0.08%,而安慰剂和萘普生的发生率为0[17]。
4.3.4 超敏性副作用
患者使用塞来昔布鲜见严重的皮肤副反应和其他超敏反应,包括过敏反应[26]。患者有对阿司匹林和/或其他NSAIDs有皮肤和/或呼吸性不耐受病史的,对塞来昔布仅有低水平的交叉非耐受性。针对骨关节炎、风湿性关节炎患者的6个月的塞来昔布治疗关节炎长期安全性研究(CLASS)800mg/d塞来昔布比一些NSAIDs(2400mg/d布洛芬或150mg/d双氯芬酸)引起更少的消化道、肝脏、出血和肾脏副作用,但是皮疹、痒风和因皮肤反应撤出实验的发生率显著偏高[27]。
5 结 语
塞来昔布(1)是第一个选择性COX-2抑制剂,十多年来它因溃疡或上消化道综合症发生率低,而被较广泛地用于风湿性关节炎、骨关节炎及这些疾病引起的疼痛治疗,其临床需求和市场的潜力是巨大的。为此,本文归纳了有代表性的塞来昔布合成方法、晶形和杂质,并对各合成方法的优缺点作了分析与总结,显然,这方面有待解决的问题尚存,例如,怎样进一步控制和减少合成塞来昔布(1)主流方法中副产物——区域异构体20的产生;对于不形成副产物20的其他方法又如何通过工艺改进等手段,达到降低成本,使之成为合成塞来昔布(1)的主流方法。此外,塞来昔布中一些主要杂质的药理学与毒理学性质仍未见文献的报道。这些都是深入研发该类药品值得思考和解决的问题。
塞来昔布(1)的临床安全和有效性已得到证实,但对其的临床研究一直都在继续,不同剂量、不同给药方式和不同疾病组群等等因素的影响都还在评价中,塞来昔布一些新的药理活性及其机制也在研究中。我们期待这些研究结果,以能够更好地指导临床用药。
[1] Davies NM,McLachlan AJ,Day RO,et al.Clinical Pharmacokinetics and Pharmacodynamics of Celecoxib[J].Clin Pharmacokinet, 2000,38(3):225-242.
[2] Penning TD,Talley JJ,Bertenshaw SR,et al.Synthesis and Biological Evaluation of the 1,5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors:Identification of 4-[5-(4-Methylphenyl)-3- (trifluorome-thyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635,Celecoxib)[J].J Med Chem,1997,40(9):1347-1365.
[3] Reddy AR,Sampath A,Goverdhan G,et al.An Improved and Scalable Process for Celecoxib: A Selective Cyclooxygenase-2 Inhibitor[J].Org Process Res Dev,2009,13(1):98-101.
[4] Reddy MVR,Bell S.C.Processes for the preparation of 1,5-diaryl-3-substituted-pyrazoles[R].Annals of Onconova Therapeutics,Inc, USA.O03024958,2003-03-27.
[5] Oh LM.Synthesis of celecoxib via 1,3-dipolar cycloaddition[J]. Tetrahedron Lett,2006,47(45):7943-7946.
[6] Beveridge RE,Fernando D,Gerstenberger BS.One-pot coppercatalyzed synthesis of N-functionalized pyrazoles from boronic acids[J].Tetrahedron Lett,2010,51(38):5005-5008.
[7] Gaulier SM,McKay R,Swain NA.A novel three-step synthesis of Celecoxib via palladium-catalyzed direct arylation[J].Tetrahedron Lett,2011,52(45):6000-6002.
[8] Ferro LJ,Miyake PJ.Polymorphic crystalline forms of celecoxib [R].Annals of Pharmacia Corporation,USA.WO0142222,2001-06-14.
[9] Vasu DR,Rekha KS,Vyas K,et al.Celecoxib,a COX-II inhibitor[R]. Acta Crystallogr C- Crys Str Commun.1999,C55,IUC9900161.
[10] Muppidi VK,Rangineni S,Duggirala NK,et al.Process for preparation of celecoxib crystalline form Annals,Dr[R].Reddy's Laboratories Ltd.,India; US2011213159,2011-09-01.
[11] Hageman MJ,He X,Kararli TT,et al.Solid-state form of celecoxib having enhanced bioavailability[R].Annals,Pharmacia Corporation,USA .US2007202160,2007-08-30.
[12] Guenduez H,Bahar M,Goektepe M.A crystalline form of celecoxib[R].Annals,Fako Ilaclari A.S.,Turk.EP1167355,2002-01-02.
[13] Ndzie E.Celecoxib forms.Annals,Generics [UK] Limited,UK. WO03091221,2003-11-06.
[14] Srinivasu MK,Narayana CL,Rao DS,et al.A validated LC method for the quantitative determination of celecoxib in pharmaceutical dosage forms and purity evaluation in bulk drugs[J].J Pharmaceut Biomed,2000,22(6):949.
[15] Satyanarayana U,Rao DS,Kumar YR,et al.Isolation,synthesis and characterization of impurities in Celecoxib a cox-2 inhibitor[J].J Pharmaceut Biomed,2004,35(4):951.
[16] Rao RN,Meena S,Nagaraju A,et al.Liquid-Chromatographic Separation and Determination of Process-related Impurities,Including a Regio-Specific Isomer of Celecoxib on Reversed-Phase C18 Column Dynamically Coated with Hexamethyldisilazane[J]. Anal Sci,2006,22(9):1257-1260.
[17] McCormack PL.Celecoxib: a review of its use for symptomatic relief in the treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis[J].Celecoxib Drugs,2011,71(18):2457-2489.
[18] McAdam BF,Catella-Lawson F,Mardini IA,et al.Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: The human pharmacology of a selective inhibitor of COX-2[J].Proc Natl Acad Sci USA,1999,96(1):272-277.
[19] Leese PT,Hubbard RC,Karim A,et al.Effects of Celecoxib,a Novel Cyclooxygenase-2 Inhibitor,on Platelet Function in Healthy Adults: A Randomized,Controlled Trial[J].J Clin Pharmacol,2000, 40(2):124-132.
[20] Simon LS,Weaver AL,Graham DY,et al.Anti-inflammatory and Upper Gastrointestinal Effects of Celecoxib in Rheumatoid Arthritis-A Randomized Controlled Trial[J].JAMA,1999,282(20): 1921-1928.
[21] Emery P,Zeidler H,Kvien TK,et al.Celecoxib versus diclofenac in long-term management of rheumatoid arthritis: randomised double-blind comparison[J].Lancet,1999,354(9196):2106-2111.
[22] Shi W,Wang YM,Li LS,et al.Safety and Efficacy of Oral Nonsteroidal Anti-Inflammatory Drugs in Patients with Rheumatoid Arthritis[J].Clin Drug Invest,2004,24(2):89-101.
[23] Bertagnolli MM,Eagle CJ,Zauber AG,et al.Celecoxib for the Prevention of Sporadic Colorectal Adenomas[J].N Engl J Med,2006, 355(9):873-884.
[24] Solomon SD,McMurray JJV,Pfeffer MA,et al.Cardiovascular Risk Associated with Celecoxib in a Clinical Trial for Colorectal Adenoma Prevention[J].N Engl J Med,2005,352(11):1071-1080.
[25] Whelton A,Maurath CJ,Verburg KM,et al.Renal safety and tolerability of celecoxib,a novel cyclooxygenase-2 inhibitor[J].Am J Ther,2000,7(3):159-175.
[26] Ltd P.Celebrex(celecoxib): summary of product characteristics[M]. Published Online,2011.
[27] Silverstein FE,Faich G,Goldstein JL,et al.Gastrointestinal Toxicity With Celecoxib vs Nonsteroidal Anti-inflammatory Drugs for Osteoarthritis and Rheumatoid Arthritis-The CLASS Study: A Randomized Controlled Trial[J].JAMA,2000,284(10):1247-1255.
Development in the Synthesis, Pharmacology, Clinical Applications of Celecoxib
YAO Shi-ning1, XU Chang2, ZHANG Qing-qing2, HUANG Hua1, YAO Qi-zheng2
(1 Market R&D Department, Eastwell International Trading Co., Ltd., Nanjiang 210002, China; 2 School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China)
Synthesis methods of Celecoxib, a COX-2 inhibitor, were reviewed and their advantages and disadvantages were described. Common crystalline forms of Celecoxib and impurities were reviewed. Pharmacology, clinical applications and adverse effect events were discussed.
Celecoxib; COX-2 inhibitor; Synthesis; Crystalline forms; Impurities; Pharmacology; Clinical applications; Side effects; Review
R96
A
1671-8194(2013)28-0051-05
*通讯作者
