合成西他沙星的研究进展*
2012-11-21陈国彬岳利剑
陈国彬, 岳利剑
(中国科学院 成都有机化学研究所,四川 成都 610041)
西他沙星(1, Chart 1)属于喹诺酮药物中的一种,由日本第一三共社于1992年开发,并于2008年在日本首次上市。1在N1-上有一个顺式氟代环丙烷取代基,与反式结构相比具有较强的抗菌活性,与不含氟的类似物相比亲油性降低[1];1对革兰阴性菌、革兰阳性菌以及厌氧菌都有很高的抗菌活性,是一种广谱抗菌药物[2,3]。对一些已经产生耐药性的致病菌,1也有很好的疗效:如Masato Touyama等[4]发现1对耐左氧氟沙星的肺炎链球菌有较好的抗菌活性;Shetty等[5]报道1对耐甲氧西林的黄色葡萄球菌以及耐万古霉素的肠球菌均有治疗作用。
含一个手性中心的螺环片段(2, Chart 1)和含邻位双手性中心的氟代环丙胺(3, Chart 1)是合成1的关键中间体,本文重点介绍合成2与3衍生物的研究进展。
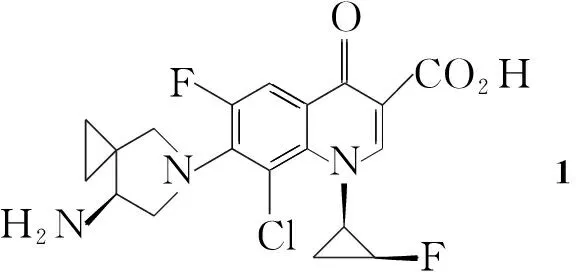
Chart1
1 2衍生物的合成
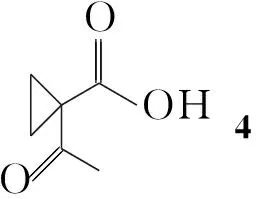
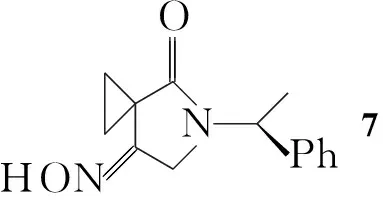
Youichi Kimura等[6]报道了2衍生物的合成方法(Scheme 1):利用乙酰乙酸乙酯与1,2-二溴乙烷反应,再水解得环丙烷羧酸化合物(4); 4与手性苯乙胺反应得5;5经溴代后关环,并与盐酸羟胺反应,然后还原肟衍生物可得8,分离两个非对应异构体可得到S-构型的产物,通过还原等步骤可得到目标产物11。
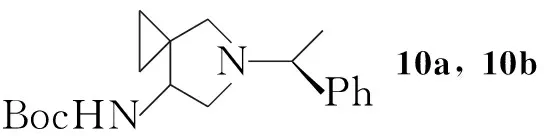
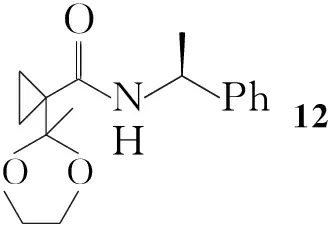
5直接溴代后在强碱条件下关环合成6的方法产率很低。为了解决这个问题,Youichi Kimura等[6]设计了另外一种合成6的方法(Scheme 2):5先与乙二醇反应形成缩酮后再进行溴代反应制得13;用氢化钠拔除酰胺氮上的氢并进攻溴原子关环得到螺环化合物后脱保护合成6。与Scheme 1相比虽然反应步骤增加,但是6的产率提高至45%(以4计算)。
Scheme1
Scheme2
Scheme3
Scheme1路线已经在1的合成工艺中规模化应用,通过手性拆分11的ee值在98%以上,但其拆分收率低,成本较高。
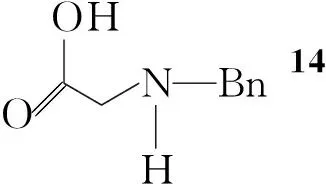
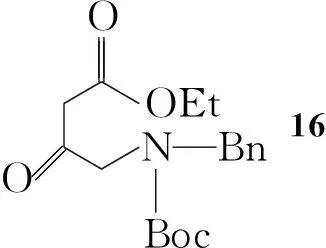
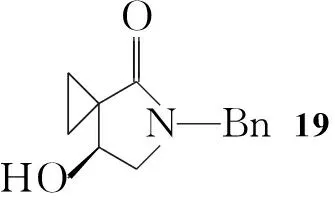
Koji Satoh等[7]报道了一种利用微生物参与羰基不对称还原的合成路线(Scheme 3):以苄基保护的氨基酸(14)为起始原料,用Boc保护后与丙二酸单乙酯反应得16;16与1,2-二溴乙烷反应,在三氟乙酸/甲苯回流的条件下关环得18,再利用微生物还原得19,其ee值大于98%。将19的羟基转变为胺后通过后续反应得到S-11。
Scheme 3路线没有利用手性拆分,而是用微生物(phaeocreopsis sp.JSM 1880)催化还原。微生物对反应条件有较高的要求,特别是pH值的影响,当pH=6.0时产率可达65%, 约 98%ee。而当pH值改变时,产率会大幅度下降,选择性也不好,如当pH=8.0时,产率仅为20%。因此Scheme 3路线在大规模生产中势必会对设备及反应条件提出较高的要求。
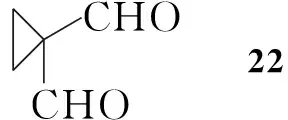
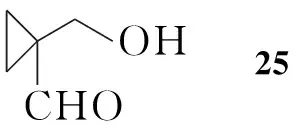
Scheme4
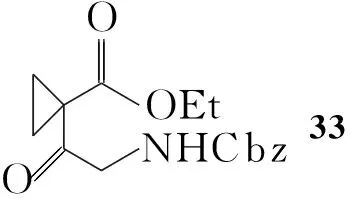
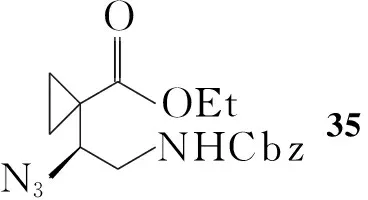
Scheme5
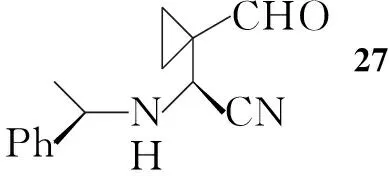
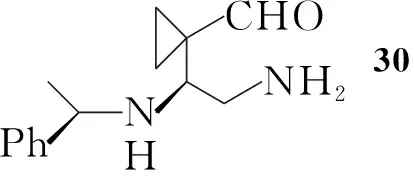
Nakayama等[8]以丙二醛为起始原料,与1,2-二溴乙烷反应生成22;22与原甲酸三乙酯反应得23;23用氢化铝锂还原,接着水解得25;25与手性苯乙胺反应生成亚胺并进一步与氰化钾反应得26,拆分后得到构型单一的产物。经氧化,再与原甲酸三乙酯反应得28;28还原氰基、除保护基后发生分子内缩合反应得到亚胺化合物31;31还原得32,进一步反应得2(Scheme 4)。Scheme 4与Scheme 1和Scheme 3相比,合成步骤较长,同时在反应中还需用到氰化钾,危险性较大,但所用的反应试剂相对比较简单,因此有利于放大生产。
Zhaoguo Zhang等[9]最近报道了一种新的合成2的方法(Scheme 5):利用铑配合物催化羰基的不对称氢化,不需用手性苯乙胺拆分,而是在由33合成34时用金属铑催化羰基进行不对称氢化反应得34,重结晶后ee值可达99%。然后用叠氮取代羟基,并对叠氮衍生物进行还原后脱除保护基,关环得36。用四氢化锂铝还原,最终得到2的盐酸盐,ee值达98%。
Scheme 5路线避免了拆分,减少了原料的浪费,并且2的ee值较高,但要使用贵金属铑,对反应条件要求较为苛刻的膦配体以及危险性较高的叠氮盐,这很可能限制其工业化应用,同时也需要解决金属的循环利用问题。
2 3衍生物的合成
3含两个手性碳并且处于相邻的位置,合成中常利用手性苯乙胺进行手性拆分。由于3是邻位双手性中心的三元环结构,其高选择性的不对称合成未见报道。
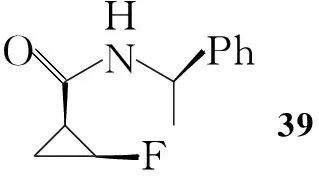
Hayakawa等[10]以1,3-丁二烯为起始原料,经四步反应合成了环丙烷羧酸酯(37),分离出顺式消旋体后,经水解并与手性苯乙胺反应得到非对应异构体39,进行拆分得到目标构型的39,水解后得到立体构型单一的氟代环丙烷羧酸(38),最后反应得到Boc保护的(1R,2S)-氟代环丙胺(40, Scheme 6)。
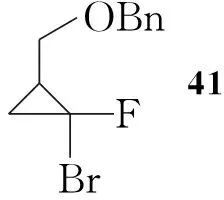
Yukimoto等[11]报道了以烯丙基苄基醚为起始原料合成3衍生物的方法(Scheme 7):烯丙基苄基醚与二溴氟甲烷环化得到氟溴取代的41,脱去苄基后得42;42再用Zn还原消除溴得43,分离得到顺式产物,用高锰酸钾氧化43得38,然后与手性苯乙胺反应得39,通过拆分可得到立体构型单一的化合物,其后续反应与Scheme 6类似。
Scheme 6与Scheme 7有相似之处,都利用了二溴氟甲烷形成的卡宾与烯烃环化得到三元环衍生物,然后用不同的方法消除溴原子,并且用高锰酸钾氧化得到羧酸衍生物。为了得到(1R,2S)-3都必须通过分离顺反异构体和不对称拆分,导致总产率降低,对原料浪费大,成本高。
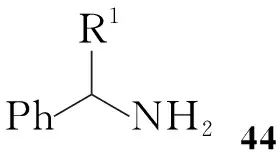
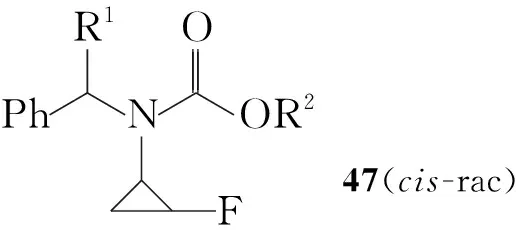
在Shiro Terashima等[12,13]报道的方法中,合成开始时先引入手性胺,通过与乙醛反应后接着与二聚光气反应得45;45与醇反应,以及环丙烷化反应后分离得到顺式的47,催化氢化得到消旋的顺式3的盐酸盐,接着用L-氯甲酸薄荷醇酯拆分最终可得到(1R,2S)-3(96%ee, Scheme 8)。
Scheme 8同样需要拆分才能得到立体构型单一的化合物,不同之处在于合成开始便引入手性胺,因此不需要将氟代环丙烷衍生物转变为羧酸后再与手性试剂反应进行拆分,这样可以较大地缩短反应步骤。由46合成47有较好的顺式选择性,通过改变R1和R2两基团,顺式与反式的比例可以达到93 ∶7。该路线步骤虽然缩短,但最后拆分顺式-3的方法相对比较繁琐。
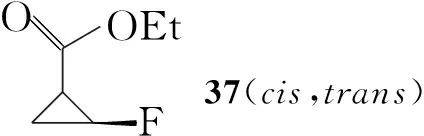
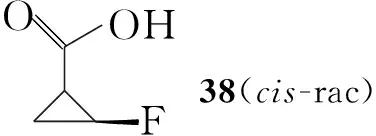
Scheme6
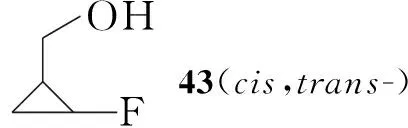
Scheme7
R1=Me or Ph, R2=Bn or Bu
Scheme8
Scheme9

Scheme10
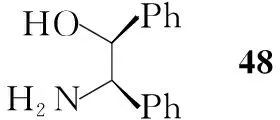
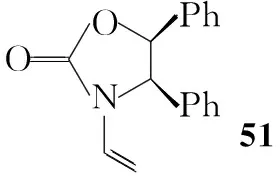
Shiro Terashima等[14,15]报道了利用手性噁唑酮作为辅助基团进行不对称合成3的方法(Scheme 9)。该路线中利用手性氨基醇(48)与二聚光气反应,然后与乙醛缩二甲醇反应得50,加热消除甲醇得51,环化得52。52含四个非对应异构体,其分离比较容易。对环丙烷化反应的反应条件进行筛选可以提高目标构型的产率,如以DCM为溶剂,加入DME和4 Å分子筛,目标构型的产率可以达到65%,分离得到立体构型单一的产物后催化氢化并进一步反应得40。
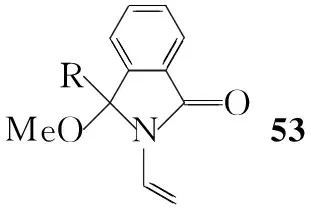
Junichi Matsuo等[16]报道了以53合成cis-3的方法(Scheme 10):首先53在碱性条件下与二溴氟甲烷发生环化反应,然后还原消除溴得54,通过改变R基团,cis∶trans可以达到84 ∶16,分离出顺式异构体后脱去羟基保护基,用硼氢化钠还原得56,在对甲苯磺酸存在下反应得到cis-3的对甲苯磺酸盐。该合成路线在环丙烷化反应中有较好的选择性,可得到顺式为主的产物,但要得到(1R,2S)-3同样也需要进行手性拆分。
从Scheme 10可以看出,由于3有两个手性碳并且处于相邻的位置,这给不对称合成造成了困难。在上述的合成路线中,Scheme 9的方法实现了用手性辅助基团控制的不对称合成,虽然目标构型的产率还不高,但仍有改进的空间。Scheme 8和Scheme 10的方法实现了选择性的合成顺式消旋体,减少了分离顺反异构体造成的浪费,但最终要想得到(1R,2S)-3都需要进行不对称拆分。而其它路线都合成了消旋体,然后拆分得到立体构型单一的氟代环丙胺,导致总收率下降。这在工业化生产中势必会造成很大的浪费,也使得成本大量增加。
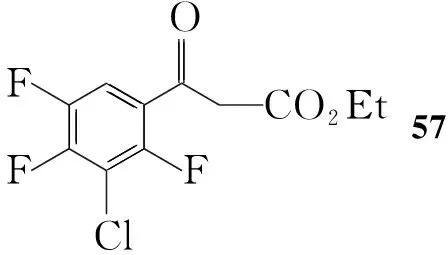

Scheme11
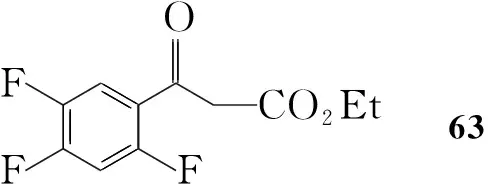
Scheme12
3 1的合成
通过三个片段的连接即可合成1。Scheme 11[6]以57为起始原料与原甲酸三乙酯反应后,再与40反应得59;59在强碱作用下得60;60水解后与2的衍生物11反应得62,最后脱保护基制得1。
也可以63为起始原料,通过类似方法合成64,再利用二氯亚砜在苯环上引入氯原子得62;62脱保护制得1(Scheme 12)[17]。
综上所述,我们对1的合成方法进行了回顾,从1的发现到现在为止很多合成方法已有报道,不对称合成2已经实现,而高选择性的合成3仍是一个挑战。
对1的药效研究也在不断深入,如Murakami等[18]首次发现1对幽门螺旋杆菌有较强的抑制作用。由于1药效强,毒副作用较小,因此有着广泛的应用前景,如何更高效地合成1,特别是不对称合成含有一个手性中心的2和含有邻手性的3将更为重要。避免因手性拆分造成的原料浪费,缩短合成路线以及提高目标产物的ee值将是今后合成1的研究重点。同时我们课题组最近在3的合成中取得了一定的进展,准备申请专利并予以报道。
[1] Atarashi S, Imamura M, Kimura Y,etal. Fluorocyclopropyl quinolones.1.Synthesis and structure-activity relationships of 1-(2-fluorocyclopropyl)-3-pyridonecarboxylic acid antibacterial agents[J].Journal of Medicinal Chemistry,1993,36(22):3444-3448.
[2] Sato K, Hoshino K, Tanaka M,etal. Antimicrobial activity of DU-6859,a new potent fluoroquinolone,against clinical isolates[J].Antimicrob Agents Chemother,1992,36(7):1491-1498.
[3] Nakajima R, Kitamura A, Someya K,etal. In vitro and in vivo antifungal activities of DU-6859a,a fluoroquinolone,in combination with amphotericin B and fluconazole against pathogenic fungi[J].Antimicrob Agents Chemother,1995,39(7):1517-1521.
[4] Touyama M, Higa F, Nakasone C,etal. In vitro activity of sitafloxacin against clinical strains of Streptococcus pneumoniae with defined amino acid substitutions in QRDRs of gyrase A and topoisomerase Ⅳ[J].Journal of Antimicrobial Chemotherapy,2006,58(6):1279-1282.
[5] Shetty N, Wilson A P R. Sitafloxacin in the treatment of patients with infections caused by vancomycin-resistant enterococci and methicillin-resistant staphylococcus aureus[J].Journal of Antimicrobial Chemotherapy,2000,46(4):633-638.
[6] Kimura Y, Atarashi S, Kawakami K,etal. (Fluorocyclopropyl)quinolones.2.Synthesis and stereochemical structure-activity relationships of chiral 7-(7-amino-5-azaspiro[2.4]heptan-5-yl)-1-(2-fluorocyclopropyl)quinolone antibacterial agents[J].Journal of Medicinal Chemistry,1994,37(20):3344-3352.
[7] Satoh K, Imura A, Miyadera A,etal. An efficient synthisis of a key intermediate of DU-6859a via asymmetric microbial reduction[J].Chem Pharm Bull,1998,46(4):587-590.
[8] Nakayama K, Muto M, Saito T,etal. Processes for preparation of bicyclic compounds and intermediates therefor[P].PC 2 002 014 278,2002.
[9] Yao Y, Fan W, Li W,etal. Synthesis of (S)-7-amino-5-azaspiro[2.4]heptane via highly enantioselective hydrogenation of protected ethyl 1-(2-aminoaceto)cyclopropanecarboxylates[J].The Journal of Organic Chemistry,2011,76(8):2807-2813.
[10] Hayakawa I, Atarashi S, Kimura Y,etal. In 31st Interscience Conference on Antimicrobial Agents and Chemotherapy[C].Chicago,1991,Abstract No.1504:Hayakawa I, Kimura Y, Japan Kokai Tokkyo Koho,1990:JPZ231475.
[11] Yukimoto J, Ehata T, Tojo T. Preparation of 2-fluorocyclo propanemethanol and 2-fluoro cyclopropanecarboxylic acid[P].JP 07 109 237,1995.
[12] Tamura O, Hashimoto M, Kobayashi Y,etal. Synthesis and optical resolution ofdl-cis-2-fluorocyclo propylamine,the key component of the new generation of quinolonecarboxylic acid,DU-6859[J].Tetrahedron Letters,1992,33(24):3483-3486.
[13] Tamura O, Hashimoto M, Kobayashi Y,etal. Synthetic studies on the key component of the new generation of quinolonecarboxylic acid,DU-6859.1.Synthesis of (1R,2S)-2-fluorocyclopr opylamine by the use of optical resolution[J].Tetrahedron,1994,50(13):3889-3904.
[14] Tamura O, Hashimoto M, Kobayashi Y,etal. Asymmetric synthesis of (1R,2S)-2-fluorocyclopropylamine,the key intermediate of the new generation of quinolonecarboxylic acid,DU-6859[J].Tetrahedron Letters,1992,33(24):3487-3490.
[15] Akiba T, Tamura O, Hashimoto M,etal. Synthetic studies on the key component of the new generation of quinolonecarboxylic acid,DU-6859.2.Asymmetric synthesis of (1R,2S)-2-fluorocyclopropylamine[J].Tetrahedron,1994,50(13):3905-3914.
[16] Matsuo J, Tani Y, Hayakaway Y. Synthesis ofcis-2-fluorocyclopropyl amine by stereoselective cyclopropanation under phase-transfer conditions[J].Chemistry Letters,2004,33(4):464-465.
[17] Yukimoto Y, Kaneuchi T, Kimura Y. Process for preparing 8-chloro quinolone derivatives[P].CN 1 062 906,1992.
[18] Murakami K, Okimoto T, Kodama M,etal. Sitafloxacin activity against helicobacter pylori Isolates,including those with gyrA mutations[J].Antimicrob Agents Chemother,2009,53(7):3097-3099.
