α-葡聚糖酶在毕赤酵母中的组成型表达*
2012-01-24梁达奉黄曾慰曾练强蚁细苗李雨虹余林
梁达奉 黄曾慰 曾练强 蚁细苗 李雨虹 余林
(1.广东工业大学轻工化工学院,广东广州510006;2.广州甘蔗糖业研究所广东省甘蔗改良与生物炼制重点实验室,广东广州510316)
甘蔗如受到刀伤或压伤、病虫害、火烧、霜冻或在收割后放置长时间,就会受到肠膜明串珠菌等微生物的感染而形成α-葡聚糖.从蔗汁澄清到糖的精炼,α-葡聚糖的存在给制糖工艺过程带来不良影响,主要体现在糖分损失、虚假旋光度的出现、糖液黏度增加、过滤困难、结晶不正常以及糖产品适用性受限制等.利用α-葡聚糖酶(EC 3.2.1.11)在制糖工艺过程分解而直接去除α-葡聚糖是目前最佳的选择:一方面,加入α-葡聚糖酶后,可消除α-葡聚糖对制糖工艺的负面影响,糖分回收提高3% ~5%,糖产品质量明显提高,使用任何其它澄清剂和化学助剂都无法达到此效果;另一方面,由经过认定的微生物产生的 α-葡聚糖酶是安全、无毒害的环保产品[1-3].
目前商品化的 α-葡聚糖酶主要是从青霉属(Penicillium)和毛壳属(Chaetomium)等微生物中得到.由于真菌产酶的同时还会产生抗生素和其它有害代谢物,成为α-葡聚糖酶通过美国食品和药物管理局(FDA)认证的一个障碍.国内外也有报道利用基因工程方法表达α-葡聚糖酶的研究,尤其是毕赤酵母表达系统,但报道的毕赤酵母工程菌均为甲醇诱导型.在甲醇诱导型毕赤酵母工程菌产外源α-葡聚糖酶的研究中,Roca等[4]通过5 L发酵罐培养获得了接近3000 U/mL的酶活.近年来,Chen等[5]用5L发酵罐培养获得了83.9 U/mL的酶活,Kang等[3]用10L发酵罐取得了134 U/mL的酶活.甲醇诱导型的醇氧化酶(AOX1)启动子作为一种在研究和生产中广泛应用的启动子[6-7],虽然具有高表达的特性和较成熟的发酵工艺,但是仍存在以下两个不足之处:(1)甲醇为易燃易爆物,给安全生产带来隐患;(2)甲醇具有毒性,作为诱导物加入发酵液后可能在产物中残留,增加了产品在食品、医药、饲料等领域应用的处理和检测成本.而pGAPZα属于组成型酵母表达载体,以三磷酸甘油醛脱氢酶(GAP)启动子作为外源基因启动子,不存在诱导的问题,在以葡萄糖或甘油等为碳源的培养基上就可以表达.研究表明该启动子的表达水平与以甲醇为诱导物的AOX1启动子的表达水平相当,是一种有巨大应用潜力的表达质粒[8-10].与 AOX1 启动子相比,GAP启动子在保持强启动子特性的同时避免了前述的两个缺陷,发酵过程无需加入诱导物即可稳定表达目标蛋白,简化了生产流程,节省人力物力.而关于组成型毕赤酵母工程菌产外源α-葡聚糖酶的研究鲜见报道.本研究采用组成型质粒pGAPZαA作为表达载体,构建了组成型表达α-葡聚糖酶的毕赤酵母工程菌,其在发酵过程中无需诱导,避免了甲醇的使用,因此提高了生产和使用安全性,降低了成本.
1 材料与方法
1.1 材料与仪器
含目的基因的载体pUC57-dex由广州甘蔗糖业研究所保存.毕赤酵母(Pichia pastoris)菌株KM71H、质粒pGAPZαA购自美国Invitrogen公司;限制性内切酶、PrimeSTAR HS DNA聚合酶、T4 DNA连接酶、E.coli JM109感受态细胞、质粒小提试剂盒和DNA凝胶回收试剂盒均购自日本Takara公司;Dextran T2000购自美国Pharmacia公司.寡核苷酸引物委托美国Invitrogen公司合成.低盐LB培养基(LLB)、YPD、YPDS培养基以及YPDS Zeocin抗性平板均按照美国Invitrogen公司的毕赤酵母表达手册配制.YPG培养基含酵母膏10 g/L、蛋白胨20 g/L、甘油30g/L.
主要仪器如下:MJ Mini型 PCR 仪、Pro-teanⅡ型蛋白电泳仪,美国BIO-RAD公司生产;DY-6C型核酸电泳仪,北京六一仪器设备有限公司生产;SKY-200B型全温度培养振荡器,上海苏坤实业有限公司生产;GEL LOGIC 200型凝胶成像系统,美国Kodak公司生产;2510型电转仪,德国Eppendorf公司生产.
1.2 表达载体pGAPZαA-dex的构建
扩增朱黄青霉(Penicillium minioluteum HI-4)基因组中的α-葡聚糖酶基因,其后连接入pUC57中得到克隆载体pUC57-dex.根据目的基因和表达载体pGAPZαA的序列特点,设计一对基因扩增引物,在上下游引物中分别引入EcoRⅠ和NotⅠ酶切位点,相应地得到CACTCTCC3'),其中下划线所示为酶切位点.以质粒pUC57-dex为模板,PCR扩增条件为:95℃ 5 min,94℃ 1min,50℃ 1 min,72℃ 2 min,共 32 个循环;72℃10 min.用胶回收试剂盒回收扩增片段.用EcoRI和NotI限制性内切酶分别对回收的基因扩增片段和质粒pGAPZαA进行双酶切,条件为37℃ 12h.酶切产物经纯化后连接过夜.连接产物转化感受态大肠杆菌,在含25μg/mL Zeocin的LLB平板上筛选阳性克隆.挑取阳性菌落扩大培养,提取质粒后分别进行聚合酶链式反应(PCR)与EcoRI、NotI双酶切鉴定.酶切与PCR鉴定正确的阳性质粒送交测序,确认序列和读码框,最后验证无误的重组质粒命名为 pGAPZαA-dex.
1.3 重组工程菌 KM71H/pGAPZαA-dex的构建
表达载体pGAPZαA-dex使用AvrⅡ于37℃进行8h酶切使之线性化.琼脂凝胶电泳检测重组质粒是否酶切完全,检测后切胶回收,纯化产物.感受态酵母细胞的制备参照文献[11].线性化质粒(10 μg)与感受态毕赤酵母KM71H(80 μL)混合,电击转化条件为:1500V、25μF、200 Ω.酵母细胞转化之后涂布于 YPDS 抗性平板上(含 100 μg/mL Zeocin),30℃培养3~4天后长出单菌落.采用1.2中所述的引物P1和P2,以煮-冻-煮法[12]制备的转化子基因组作为模板进行PCR,能够扩增得到1700 bp左右片段的转化子为阳性转化子.
1.4 产酶工程菌的摇瓶发酵
通过不同梯度浓度Zeocin的YPD平板从阳性转化子中挑选出数株高拷贝克隆.将平板上的单克隆接入50 mL YPG培养基中作为一级培养,30℃、250 r/min的条件下培养至 D(600)=5.0时,取40mL接入360mL YPG培养基中继续培养.第72小时补加甘油,甘油补加量为培养基总量的3%.每隔12h取样测定湿重、D(600)、酶活.每隔24 h收集发酵液,12000r/min离心5min,收集上清.100mg三氯乙酸(TCA)加入1mL上清液,冰浴1 h.12000 r/min离心20 min,收集沉淀,溶于 100 μL磷酸盐缓冲液(PBS),之后用聚丙烯酰胺凝胶电泳(SDS-PAGE)分析.
酶活测定方法:将重组毕赤酵母发酵液经12000r/min离心5min的上清液稀释适当倍数后即为本实验所用酶液.取900 μL 0.02 g/mL的葡聚糖(Dextran T2000)溶液,置于45℃恒温水浴中至少保温5min,然后加100μL酶液.精确反应10min,立即加入2mL二硝基水杨酸钠试剂(DNS)以终止反应,沸水浴5 min,然后迅速将其冷却,用蒸馏水定容至25mL,于540nm下测定吸光值.从标准曲线的回归方程求得相对应的葡萄糖的量,并计算出酶活.酶活定义:采用DNS法,在45℃、pH=5.5的条件下,每分钟催化底物(0.02 g/mL Dextran T2000)水解产生1μmol葡萄糖所需的酶量为1个酶活单位,以U表示.
2 结果与分析
2.1 表达载体pGAPZαA-dex的鉴定
以P1和P2作为引物通过PCR方法扩增α-葡聚糖酶基因,10g/L琼脂凝胶电泳(见图1)显示,得到单一DNA条带,长度约为1700bp,与设计的目的片段长度相符.
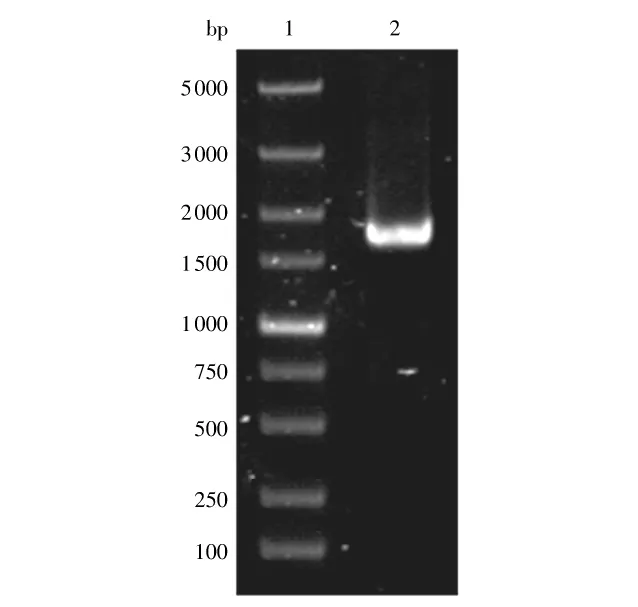
图1 PCR扩增α-葡聚糖酶基因Fig.1 PCR amplification of dextranase gene
如图2所示,α-葡聚糖酶基因经过PCR扩增后,与组成型分泌表达载体pGAPZαA连接,构建成重组质粒pGAPZαA-dex,目标基因之后连接有6×His标签的基因序列.
将连接好的表达载体进行PCR扩增鉴定,继而进行EcoRⅠ和NotⅠ双酶切鉴定,结果见图3.将该阳性质粒送交Invitrogen公司测序,结果显示基因序列完整,无碱基突变,且重组表达载体的读码框正确,表达载体pGAPZαA-dex构建成功.
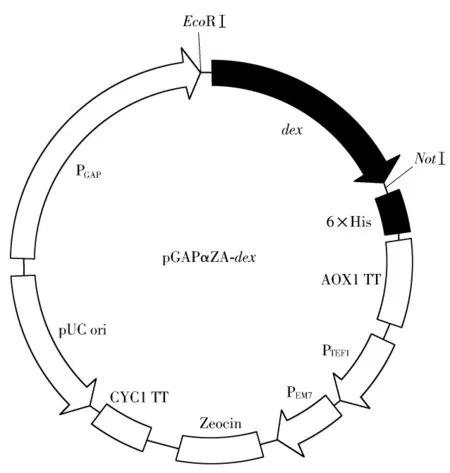
图2 重组质粒pGAPZαA-dex的结构Fig.2 Structure of recombinant plasmid pGAPZαA-dex
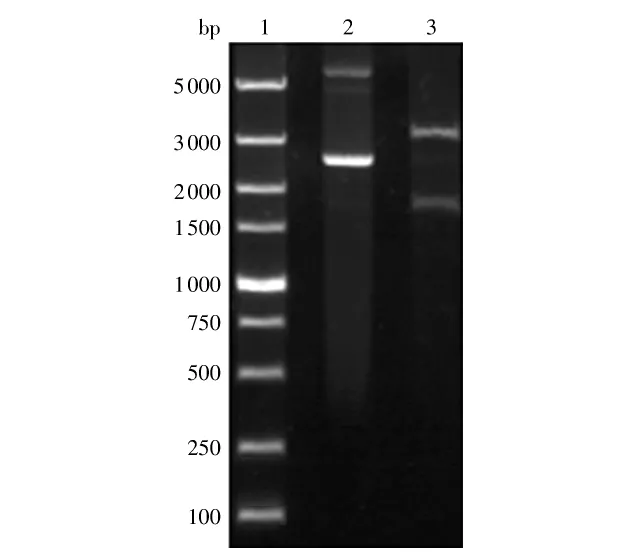
图3 质粒pGAPZαA-dex的双酶切鉴定Fig.3Characterization of pGAPZαA-dex by EcoRⅠ/NotⅠdigestion
2.2 重组工程菌的PCR鉴定
挑选若干Zeocin抗性平板上的转化子,采用煮-冻-煮法制备各转化子的基因组作为PCR检测的模板,PCR结果如图4所示.选出鉴定为阳性的转化子进行摇瓶发酵实验.
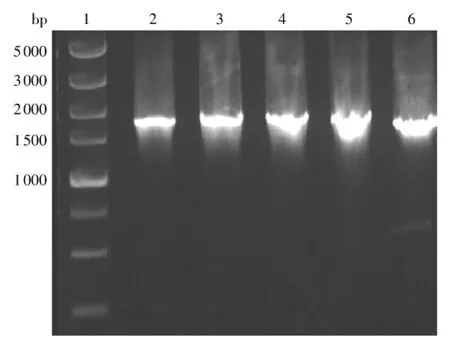
图4 经pGAPZαA-dex转化后KM71H的PCR验证Fig.4 Confirmation of KM71H transformed with pGAPZαA-dex by PCR
2.3 摇瓶发酵结果
挑选2.2摇瓶发酵实验中较高产的菌株在YPG培养中基摇瓶发酵,培养120h酶活达到60.4U/mL,菌体D(600)为38.7.接种12h之后菌体生长迅速,进入对数期.产酶量也随之较快地积累.培养72 h之后,酵母生长减缓,酶活积累也随之放缓.从发酵数据可知,72h时酶活达到50.4 U/mL,96 h时酶活达到58.5 U/mL,直至120 h达到顶点(见图5).实际上72 h至96 h之间即可作为发酵的终点取得最佳的效益.笔者正在进行的5 L发酵罐实验也验证了这一点.从酶活曲线和菌体密度曲线来看,二者不仅有较强的趋势一致性,而且还具有正相关性,说明发酵液中菌体密度越高,目标产物的浓度越高;这是组成型表达的一个显著特点.
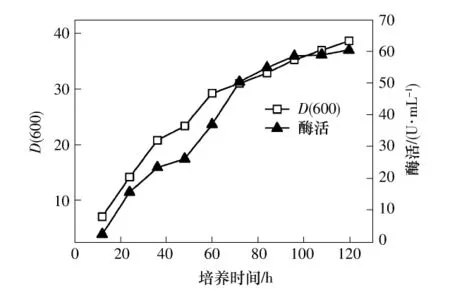
图5 摇瓶发酵酶活和D(600)变化曲线Fig.5 Dextranase activity and D(600)profiles in shake flask cultures
分析预测 α-葡聚糖酶的相对分子质量为65000,SDS-PAGE分析显示发酵液上清中含有相对分子质量接近66000的特异条带,这说明本研究成功表达了α-葡聚糖酶(见图6).
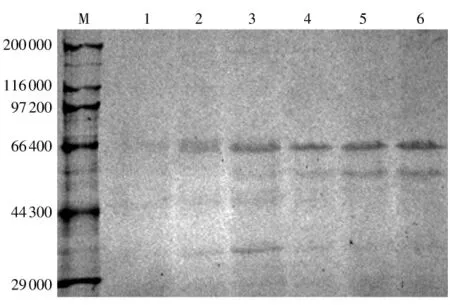
图6 摇瓶发酵液上清SDS-PAGE分析Fig.6 SDS-PAGE analysis of supernatants from shake flask cultures
3 结语
本研究构建了组成型表达载体pGAPZαA-dex,以毕赤酵母为平台成功表达了α-葡聚糖酶,该融合蛋白相对分子质量约为66000,摇瓶发酵120h酶活达60.4U/mL.组成型毕赤酵母工程菌的发酵避免了有毒易燃易爆品——甲醇的使用,既便于大规模生产,也有利于发酵产品通过食品药品安全认证,为探索适用于制糖工业的α-葡聚糖酶的开发打下了基础.下一步将进行发酵罐流加培养工程菌的试验,逐级放大培养规模,通过优化发酵工艺和分离纯化工艺,降低α-葡聚糖酶的生产成本.
[1] Eggleston G,Monge A,Montes B,et al.Application of dextranases in sugarcane factory:overcoming practical problems[J].Sugar Tech,2009,11(2):135-141.
[2] Eggleston G,Mongeb A.Optimization of sugarcane factory application of commercial dextranases [J].Process Biochemistry,2005,40(5):1881-1894.
[3] Kang H K,Park J Y,Ahn J S,et al.Cloning of a gene encoding dextranase from Lipomyces starkeyi and its expression in Pichia pastoris[J].Journal of Microbiology and Biotechnology,2009,19(2):172-177.
[4] Roca H,Garcia B,Rodriguez E,et al.Cloning of the Penicillium minioluteum gene encoding dextranase and its expression in Pichia pastoris[J].Yeast,1996,12(12):1187-1200.
[5] Chen L,Zhou X,Fan W,et al.Expression,purification and characterization of a recombinant Lipomyces starkeyi dextranase in Pichia pastoris[J].Protein Expression and Purification,2008,58(1):87-93.
[6] 罗立新,黄志立,潘力,等.纳豆激酶基因在巴斯德毕赤酵母中的表达[J].华南理工大学学报:自然科学版,2003,31(2):1-4.Luo Li-xin,Huang Zhi-li,Pan Li,et al.Expression of nattokinase gene in yeast Pichia Pastoris[J].Journal of South China University of Technology:Natural Science Edition,2003,31(2):1-4.
[7] Macauley-Patrick S,Fazenda M L,McNeil B,et al.Heterologous protein production using the Pichia pastoris expression system [J].Yeast,2005,22(4):249-270.
[8] Gasser B,Maurer M,Gach J,et al.Engineering of Pichia pastoris for improved production of antibody fragments[J].Biotechnology and Bioengineering,2006,94(2):353-361.
[9] Pal Y,Khushoo A,Mukherjee K J.Process optimization of constitutive human granulocyte-macrophage colony-stimulating factor(hGM-CSF)expression in Pichia pastoris fed-batch culture[J].Applied Microbiology and Biotechnology,2006,69(6):650-657.
[10] Zhao W,Wang J,Deng R,et al.Scale-up fermentation of recombinant Candida rugosa lipase expressed in Pichia pastoris using the GAP promoter[J].Journal of Industrial Microbiology and Biotechnology,2008,35(3):189-195.
[11] Goodrick J C,Xu M,Finnegan R,et al.High-level expression and stabilization of recombinant human chitinase produced in a continuous constitutive Pichia pastoris expression system [J].Biotechnology and Bioengineering,2001,74(6):492-497.
[12] 剧海,梁东春,郭刚,等.用于PCR实验的毕赤酵母基因组DNA制备方法的比较[J].天津医药,2003,31(5):270-272.Ju Hai,Liang Dong-chun,Guo Gang,et al.Comparison of four methods to prepare Pichia genomic DNA for PCR[J].Tianjin Medical Journal,2003,31(5):270-272.
