HPLC法测定云南松松针中莽草酸的含量
2011-04-17孔艳飞李爱玲赵振华杨梅飞
王 琳 孔艳飞 李爱玲 赵振华 杨梅飞
云南省大理学院药学院,云南 大理 671000
松针 (pine need1e)又名松叶,为松科松属 (Pinus)植物马尾松、华山松、黄山松、黑松、油松、云南松、红松等的针叶,俗称松毛、山松须、猪鬃松叶等。 《本草纲目》曰:“久服令人不老,轻身益气,主治风湿疮,生毛发,安五脏,守中,不饥延年。”[1]针叶具有活血、祛风,燥湿止痒之功,可用于治疗风湿疥癣等疾病[2]。现代药理学研究证明,莽草酸具有较强的抗炎、镇痛和抑制血小板聚集作用[3-4],也是目前临床上唯一对感染禽流感病毒患者有效药物“达菲”的基本原料[5]。目前有关莽草酸的化学合成,由于反应条件苛刻、成本高而难于满足市场需求,因此筛选价廉的莽草酸提取原料显得十分迫切[6]。我国云南松资源丰富,为了寻找该植物中的活性成分,综合利用其资源,使其变废为宝,本实验采用HPLC法测定云南松松针中莽草酸的含量,为有针对性的开发利用我国各地丰富的云南松资源中莽草酸提供科学依据。
1 仪器与试药
Agi1ent 1200型高效液相色谱仪 (包括四元泵,自动进样器,DAD检测器,chemstation化学工作站);SY3200-7型超声波清洗器 (上海声源超声仪器设备有限公司);AE240电子天枰 (梅特勒一托利多仪器 (上海)有限公司)。
莽草酸对照品 (成都曼斯特生物科技有限公司,HPIC测定,归一化法计算其纯度≥99%,批号A0112)。甲醇为色谱纯 (美国Tedia公司),水为娃哈哈牌纯净水,其它均为国产分析纯试剂。
云南松样品为2010年2月采集于云南省大理市漾濞县,经马晓匡教授鉴定为云南松 (pinus yunnanensis Franch)。
2 色谱条件
色谱柱为Agi1ent Ec1ipse XDB-C18(4.6mm ×150mm,5μm),流动相为甲醇-1%磷酸水溶液 (3:97),流速为0.8m1·min-1,检测波长为 214nm,柱温为 25℃。
3 系统适用性实验
3.1 理论塔板数 (n) 按莽草酸峰计算为7766.428,不低于5000。
3.2 分离度 与其它峰的分离度为2.1,均大于1.5;对照品溶液和供试品溶液分别进样10μ1,记录色谱图,莽草酸的保留时间约为2.8min,见图1。
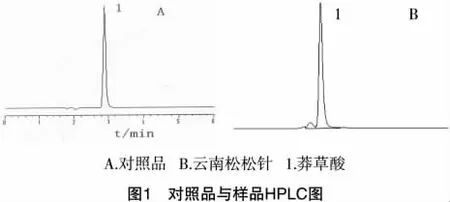
4 分析方法认证与结果
4.1 溶液的制备
4.1.1 对照品溶液的制备 精密称取莽草酸对照品0.0078g(7.8mg),置于100m1容量瓶中,加重蒸水溶解并定容至刻度,摇匀,即得对照品贮备液 (每1m1含莽草酸78ug)。
4.1.2 供试品溶液的制备 精密称取云南松松针0.1003g,置于100m1具塞锥形瓶中,加重蒸水60m1,超声处理 (功率200W,频率33kHz)90min,过滤,定容至100m1,临用前用0.45μm微孔滤膜过滤并装入进样瓶中,得云南松松针供试品溶液。
4.2 标准曲线的制备 精密吸取莽草酸对照品贮备液1,2,4,6,8,10,12,14m1,分别置于25m1容量瓶中并定容,浓度 分 别 为 3.12 ug/m1,6.24 ug/m1,12.48 ug/m1,18.72 ug/m1,24.96 ug/m1,31.20 ug/m1,37.44 ug/m1,43.68ug/m1,摇匀,用0.45um微孔滤膜过滤并分别装入进样瓶中备用。分别精密吸取10u1注入色谱仪,得进样量C(ug)与峰面积A的线性方程。结果表明,莽草酸进样量在0.0312~0.4368ug与峰面积成良好的线性关系,回归方程为 A=5042.6C+28.2490,r=0.9999(n=8)。
4.3 重复性试验 精取莽草酸对照品溶液 (31.20ug/m1)10u1,重复进样 5次,测得色谱峰峰面积平均值为1602.0859,RSD为0.037%,符合分析要求。
4.4 精密度试验 精密称取同一批云南松松针干燥粉末6份,按样品测定法重复测定6次,测得莽草酸含量的平均值为1.34%,RSD为3.04%,符合分析要求。
4.5 稳定性试验 取云南松松针供试品溶液,在“2”项色谱条件下,于0、4、8h分别测定莽草酸峰面积,计算峰面积的RSD为0.54%,表明供试液在8h内稳定性良好。
4.6 准确度实验 (加样回收试验) 精密称取已知含量的云南松松针粉末约0.1g,共6份,分别精密加入一定量的莽草酸对照品,按供试品溶液制备及测定法操作,分别进行色谱分析,其平均回收率为99.97%,RSD为1.06%,符合分析要求。结果见表1

表1 莽草酸加样回收率(n=6)
5 讨论
5.1 测定方法的选择 目前,国内外对莽草酸的测定有正相色谱法、离子抑制反相色谱法和毛细管电泳法等。虽然这些方法可以测定莽草酸,但是存在不足之处:使用正相色谱时,不但要用专用正相色谱柱而且流动相大部分是乙腈,运行成本较高;离子抑制反相色谱法使用PH值很低的酸水,对普通ODS柱长期使用会损坏填料;毛细管电泳法是一种较新的检测方法,但其重现性较差。本试验采用高效液相色谱法测定莽草酸含量,实验结果符合莽草酸含量测定要求,且运行成本低。
5.2 供试品溶液的制备方法的选择 试验中分别考察了超声提取、加热回流、索氏提取等方法的提取效果,以超声提取法最为省时,提取效率最高。而超声提取法中,又分别比较了不同的溶剂 (重蒸水、甲醇、95%乙醇)、不同超声时间、溶剂不同体积倍数的提取效果,试验表明以60倍量的重蒸水为提取溶剂,超声90min,可将莽草酸基本提取完全[7]。
5.3 流动相的选择 实验比较了甲醇-水、乙腈-水等流动相系统[8,9],从分离情况和出峰时间等综合分析选择甲醇-1%磷酸水溶液等度洗脱为佳,且保留值适宜,柱后处理简便、省时。
5.4 检测波长的选择 用二极管阵列检测器在本试验溶剂体系条件下,分析了莽草酸对照品色谱峰和样品中目标组分相应色谱峰的紫外光谱,结果保留值相同处紫外光谱基本一致,莽草酸在214nm处有最大吸收。待测组分与杂质峰基本能基线分离,检测灵敏度高,确定莽草酸检测波长为214nm。
目前,莽草酸主要来源于八角属植物八角茴香等果实中,但资源十分有限,无法满足市场的需求[10]。实验结果表明云南松松针中莽草酸含量较高,且云南松松针是云南松的主要副产物,再生速度快,且可持续利用。云南松松针的试验方法及试验结果有对莽草酸含量的研究提供了依据,也表明了云南松松针中莽草酸的开发将有巨大的潜力。
[1]李时珍.本草纲目·下册[M].北京:人民卫生出版社,1982:19~17.
[2]孙学龙,李铣.松针的研究进展[J].沈阳药学院学报,1990,7(3):224~226.
[3]马怡,孙建宁,徐秋萍,等.莽草酸对血小板聚集和凝血的抑制作用[J].药学学报,2000,35(1):1~3.
[4]方玉珍,宋杰云,岑燕飞,等.毒八角酸的镇痛作用研究[J].贵阳中医学院学报,1989,1:59~62.
[5]刘永友,廖晓峰.莽草酸的研究进展[J].化工时刊,2007,27(3):54~57.
[6]刘相奎,王强,袁哲东.莽草酸合成路线图解[J].中国医药工业杂志,2006,37(1):70~72.
[7]刘光明,周浓,夏从龙,等.HPLC法测定云南松中莽草酸的含量[J].大理学院学报,2010,9(2):14~18.
[8]汪红,边清泉.雪松叶中莽草酸的RP-HPLC测定[J].中国医药工业杂志,2007,38(11):797~799.
[9]马廉举,刘新.反相离子对色谱法测定马尾松松针中莽草酸的含量[J].中药材,2008,31(1):63~65.
[10]王晓强,郭亚健,杨春澍.高效液相法测定八角属部分植物果实中莽草酸的含量[J].中国中药杂志,2001,26(7):447~449.
