脊髓小脑性共济失调Ⅲ型的临床特点及分子遗传学特征
2010-07-04田增民亓树彬
陈 涛,田增民,亓树彬
脊髓小脑性共济失调(spinocerebell ataxias,SCAs)属于常染色体显性共济失调,除SCAs外,还包括发作性共济失调(episodic ataxia,EA)1型和2型(即EA1和EA 2),是遗传性共济失调的主要类型[1]。SCAs占神经系统遗传性疾病的 10%~15%,患病率为1~4/10万,可见于各种族[2-3]。脊髓小脑性共济失调Ⅲ型(SCA3)是亚洲人种最常见的脊髓小脑性共济失调亚型。
1 资料与方法
1.1 调查对象 一组SCA3家系8例患者,均为汉族,其中男性6例,女性2例,男女比例 3∶1。通过系谱调查,该家系呈常染色体显性遗传,且存在遗传早现现象。
1.2 方法
1.2.1 聚合酶链反应-聚丙烯酰胺凝胶电泳(PCRPAGE)技术 对8例中的3例行(CAG)n重复数目检测。
1.2.2 临床表现分析 对其中2例患者均进行全面病史询问,行神经系统检查及颅脑MRI扫描,根据结果对其临床表现进行分析。
1.2.3 家系患病情况调查 详细对患者家系询问,了解家系构成及患病情况。
2 结果
2.1 临床表现 生活自理差,无法独立行走,需搀扶,步基增宽;双眼水平、垂直震颤阳性;右侧咽反射迟钝,软腭活动度好,伸舌不偏,但构音不良,言语呈爆破音;颈软,双上肢肌张力正常,双下肢肌张力增强,四肢肌力正常,四肢腱反射(+++);Hoffmann征阳性,余病理征阴性;指鼻试验及跟膝胫试验不准,深浅感觉未见异常。辅助检查:头颅MRI示脑干、小脑体积减小,四脑室及环池扩大,小脑萎缩。
2.2 基因检查 CAG重复先证者76次,其父68次,其5个月胎儿74次,而CAG重复的正常范围为1~40次。
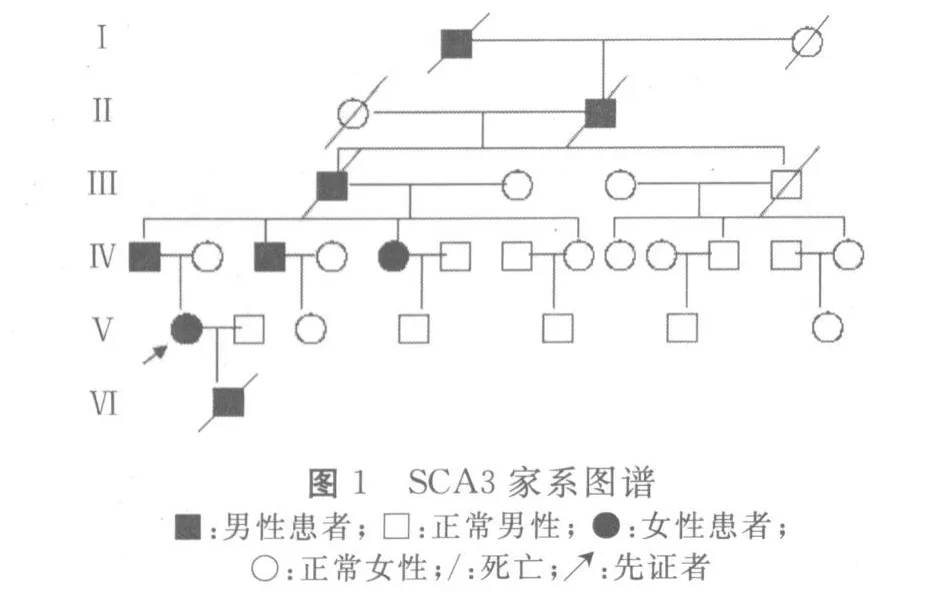
2.3 家系调查 本家系6代人中共8人患病,先证者的曾曾祖父、曾祖父、祖父、父亲、二叔及二姑均在40岁左右发病,症状同先证者类似,但症状较先证者轻,生活可自理。其中先证者、其父和其5个月龄男性胎儿已基因检测明确诊断为SCA3,其余未行基因诊断。其中先证者曾曾祖父、曾祖父和祖父已故(死亡年龄和原因不详),先证者的5个月龄男性胎儿流产。此家系已知的第5代子女的年龄在5~30岁之间,目前仅先证者发病,余尚无异常发现,其中部分亲戚已失去联系,故婚育及发病情况不详,其系图谱见图1。
3 讨论
脊髓小脑性共济失调属于常染色体显性共济失调,占神经系统遗传性疾病的10%~15%,患病率为1~4/10万,可见于各种族[2,3]。到目前为止已发现了27个与SCAs有关的基因位点(SCA1~27),其中 SCA1、SCA2、SCA3/马查多-约瑟夫病(MJD)及SCA6是最常见的亚型[4-5]。现已证实SCAs是由于其疾病基因编码区内三核苷酸(CAG)重复序列动态突变所致。由于SCAs致病基因中的三核苷酸重复动态突变,导致其编码的蛋白构象改变,从而对特异性神经元产生细胞毒性作用,并与泛素蛋白酶体通路(UPP通路)、半胱氨酸天冬酶及细胞特异性相互作用蛋白有关。本家系报道的 SCA3与 SCA1、SCA2 、SCA6 、SCA7、SCA12、SCA 17 和齿状核红核苍白球路易体萎缩(DRPLA)等8种是由于SCAs疾病基因外显子上三核苷酸CAG编码的多聚谷氨酰胺链的异常扩大,使基因产物异常聚集,从而对神经元产生毒性作用而发病,故又称多聚谷氨酰胺病(Poly Q病)[6-8]。脊髓小脑性共济失调是高度遗传异质性疾病,其基因型有以下3个特征:①早现遗传,即在CAG重复的遗传病在连续世代中,存在发病年龄一代比一代早、病情一代比一代严重的现象。(CAG)n重复数目的大小与发病年龄成反比,与发病的严重程度成正比。②在大多数SCAs中,CAG重复的数目与SCAs发病存在临界值,CAG的重复一旦超过临界值,患者就可能有临床表现。③父系遗传的病例,大都在幼年发病,进展迅速,症状严重。SCAs各亚型症状相似,交替重叠,其共同的临床表现为共济失调、眼震及构音障碍,还可有肌张力障碍、腱反射亢进及病理征阳性。除了上述共同特征,各亚型也具有各自的特点而构成不同疾病。本家系报道的SCA3在中国、日本及德国均为最常见亚型。SCAs通过典型的临床症状和体征并结合影像学检查可作出初步诊断,但确诊需靠基因诊断。本家系报道的3例患者首发症状为走路不稳、上肢乏力、震颤、饮水呛咳及言语不清,并进行性加重,MRI示脑干、小脑萎缩,临床诊断“遗传性共济失调”成立。
SCAs目前无特效治疗,传统治疗方法主要是对症治疗,如药物治疗、高压氧治疗、中医治疗、功能锻炼及康复治疗等。上述治疗可轻微缓解症状,但无法阻止病情进展。基因治疗[9]是最理想的治疗方法,但目前仍处于体外试验和动物实验阶段,用于临床还很遥远。近年来,我院开展的立体定向神经干细胞移植手术治疗小脑萎缩,是针对各种病因引起的小脑损害,其中也包括SCAs患者,并取得了一定疗效[10-11],这为SCAs患者的治疗提供了新的希望。
[1]Holton JB,Walter JH,Tyfield LA.Galactosemia[A]//Scriver CR,Beaudet A L,Sly WS,et al.The metabolic and molecular bases of inherited disease[M].8th ed.New York:McGraw-Hill Companies,2001:5741-5748.
[2]Schols L,Bauer P,Schmidt T,et al.Autosomal dominant cerebellar ataxias:clinical features,genetics,and pathogenesis[J].Lancet Neurol,2004,3(5):291-304.
[3]Manto MU.The wide spectrum of spinocerebellar ataxia(SCAs)[J].Cerebellum,2005,4(1):2-6.
[4]Orr HT,Chung MY,Banfi S,et al.Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1[J].Nat Genet,1993,4(3):221-226.
[5]Koeppen AH.The pathogenesis of spinocerebellar ataxia[J].Cerebellum,2005,4(1):62-73.
[6]唐北沙,夏家辉,王德安,等.遗传性脊髓小脑型共济失调的CAG三核苷酸突变检测[J].中华医学遗传学杂志,1999,16(5):281-284.
[7]Michlewski G,Krzyzosiak WJ.M olecular architecture of CAG repeats in human disease related transcripts[J].J Mol Biol,2004,340(4):665-679.
[8]Kawaguchi Y,Okamoto T,Taniwaki M,et al.CAGexpansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1[J].Nat Genet,1994,8(3):221-228.
[9]Miller VM,Xia H,Marrs GL,et al.Allele-specific silencing of dominant disease genes[J].Proc Natl Acad Sci USA,2003,100(12):7195-7200.
[10]田增民,李志超,尹丰,等.人神经干细胞移植治疗小脑萎缩[J].第二军医大学学报,2004,25(9):933-935.
[11]Tian ZM,Chen T,Zhong N,et al.Clinical study of transplantation of neural stem cells in therapy of inheri-ted cerebellar atrophy[J].Bejing Da Xue Xue Bao,2009,41(4):456-458.
