2例中国原发性纤毛运动障碍患者致病变异的鉴定
2024-04-26郑海霞周王继田欣伦刘雅萍
郑海霞,周王继,田欣伦*,刘雅萍
1.中国医学科学院基础医学研究所 北京协和医学院基础学院 麦库西克-张孝骞协和遗传医学中心疑难重症及罕见病国家重点实验室,北京 100005;中国医学科学院 北京协和医学院 北京协和医院2.呼吸与危重症医学科 疑难重症及罕见病国家重点实验室;3.临床医学研究所,北京 100730
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种罕见的遗传病,主要呈常染色隐性遗传,少数呈X连锁隐性遗传,其发病率因地域、人种及数据来源不同差异较大,全球范围内约为1/1万~1/4万[1-2],然而在中国尚缺乏准确的统计和报道。由于漏诊率较高,实际发病率可能高于文献报道。PCD是由动纤毛结构或功能异常引起的疾病,涉及多个器官和系统,导致包括慢性呼吸道感染、生育问题等一系列临床症状[3]。此外,约50%的PCD 患者可能伴有内脏反位,被称为Kartagener 综合征。PCD的诊断标准目前尚不统一,临床上常采用鼻呼出一氧化氮(nasal nitric oxide, nNO)水平检测、高速视频显微镜分析(high-speed video microscopy analysis, HSVA)、透射电镜(transmission electron microscope, TEM)观察和基因检测进行综合诊断[4]。PCD具有遗传异质性,目前已发现50多个致病基因,但仍有约25%的患者未找到其致病基因[5]。
本研究采用临床表型分析、全外显子组测序技术(whole exome sequencing, WES)、Sanger测序以及生物信息学分析等多种方法,对2例PCD患者的病因学展开研究,目的在于为该病的基因诊断、遗传咨询和精准治疗提供依据。
1 材料与方法
1.1 研究对象
患者1,女,37岁,曾有新生儿呼吸窘迫,自幼反复呼吸道感染。
患者2,男,32岁,10余年来反复咳嗽。
2例患者于2022年至2023年在北京协和医院呼吸内科门诊就诊。在告知患者及其家属知情同意书内容后,所有成员均签署知情同意书。咨询并记录患者病史及家族史,进行相关临床资料收集。该研究获得中国医学科学院北京协和医院伦理审查委员会的审查批准(I-24PJ0444)。
1.2 研究方法
1.2.1 样本DNA的提取:使用EDTA抗凝管采集患者外周静脉血2 mL,用QIAamp全血细胞DNA提取试剂盒(Qiagen公司)按照说明书中描述的标准方法提取基因组DNA(gDNA)。
1.2.2 全外显子组测序检测与致病变异筛选:取2 μg患者gDNA委托给诺禾致源生物科技有限公司,进行全外显子组测序和注释工作。使用ANNOVAR进行变异注释,包括千人基因组计划、ExAC数据库、gnomAD数据库、 dbSNP数据库等注释信息,注释包括变异位置、等位基因频率、变异保守性预测和致病性预测等。致病变异筛选参照以下策略:1)符合常染色体隐性或X染色体隐性遗传的遗传模式;2)最小等位基因频率≤0.01;3)位于基因编码区或可能影响剪接的位点;4)生物信息学软件预测致病性。
1.2.3 Sanger测序验证:应用Sanger测序对候选变异进行进一步验证。从University of California Santa Cruz(UCSC)基因组浏览器(https://genome.ucsc.edu/)获取基因序列,使用Primer3在变异位点附近设计引物,由北京天一辉远生物科技有限公司合成。对患者DNA样本和正常对照样本进行PCR扩增,扩增产物由北京诺赛基因组研究中心有限公司进行Sanger测序。
1.2.4 变异致病性的分析:参考人类基因突变数据库(The Human Gene Mutation Database,HGMD)数据库和相关文献的研究结果,根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)指南对候选变异位点进行致病性评估。利用Varcards网站(http://www.genemed.tech/varcards/)得到多个软件对候选变异的致病性预测(REVEL、CADD和MutationTaster等)、变异在千人基因组、ExAC及gnomAD等多个人群数据库中的频率。综合人群频率、预测致病性结果、家系共分离等证据,利用Intervar网站(https://wintervar.wglab.org/),将这些分析结果结合起来确定变异的致病性。
利用Ensembl网站(https://asia.ensembl.org/index.htmll),得到基因结构图。利用UCSC基因组浏览器对基因变异位点的保守性进行预测。
2 结果
2.1 临床表型
患者1,女,37岁,曾有新生儿呼吸窘迫,自幼反复呼吸道感染,有内脏转位,右中叶、左舌段和双下叶支气管扩张伴感染(图1左),nNO浓度均值为5 ppb(parts per billion),低于125 ppb的临界值,被诊断为Kartagener 综合征。患者不孕,试管婴儿孕育子女,子女体健。家族史:父母近亲结婚,姐姐弟弟体健。
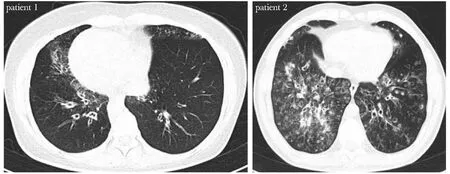
图1 2例患者的支气管扩张表型Fig1 Phenotype diagrams of the two probands with bronchiectasis
患者2,男,32岁,10余年来反复咳嗽、咯黄痰,双肺弥漫支气管扩张合并感染(图1右),nNO均值为10 ppb,低于125 ppb的临界值。不育2年。家族史:否认父母近亲结婚史,姐姐体健。
2.2 全外显子组测序分析与Sanger测序验证
分析全外显子组测序结果,并进行变异筛选,发现2例患者分别携带外动蛋白臂对接复合物亚基1(outer dynein arm docking complex subunit 1, ODAD1)(NM_144577)基因纯合变异和动力蛋白轴丝组装因子6(dynein axonemal assembly factor 6,DNAAF6)(NM_173494)半合子变异。患者1携带ODAD1:c.702_705dupGCAG (p.P236Afs*11),患者2携带DNAAF6: c.532_533delCT (p.L178Sfs*2),这2个变异均未在千人基因组、ExAC及gnomAD等公共数据库中收录,且在HGMD未见报道。Sanger测序结果表明全外显子组测序结果无误(图2)。
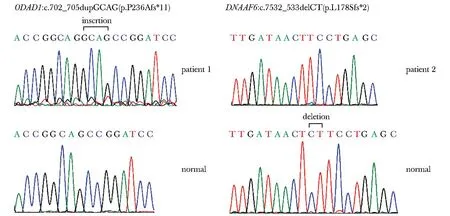
图2 2例患者与正常对照的ODAD1和DNAAF6变异位点测序图
2.3 变异致病性预测分析结果
在两例PCD患者中检出2个未被报道过的致病变异,均为移码变异。ODAD1:c.702_705dupGCAG(p.P236Afs*11)在第7号外显子插入了4个碱基后发生移码,在继续编码11个变异的氨基酸后产生提前终止密码子,导致蛋白产物缺失下游约2/3的序列(图3A)。DNAAF6: c.532_533delCT(p.L178Sfs*2)在第7号外显子上缺失了2个碱基后发生移码,提前产生终止密码子,影响了下游保守序列(图3B)。根据ACMG指南并结合文献提供的致病性判定依据进一步确认了2个新变异的致病性(表1),合并上述分级证据判定这2个新变异均为致病变异。

表1 2例患者变异致病性判定Table 1 Pathogenicity determination of the variants in 2 patients with PCD
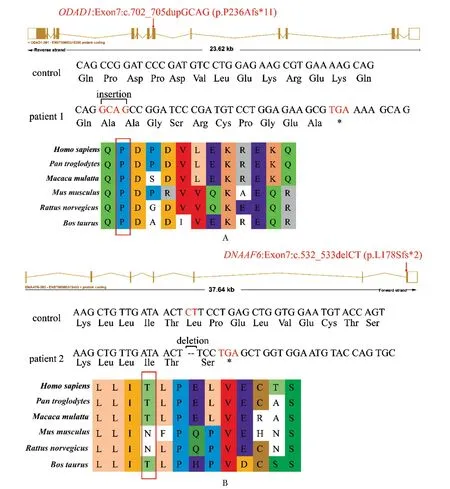
Red arrows indicated the location of the pathogenic variant identified in two patients, respectively, amino acid coding diagram from health control and two patients and conservation of the variant sites of ODAD1 gene and DNAAF6 gene (red rectangles indicated the first codon affected by the frameshift variant).
3 讨论
PCD是一种罕见的常染色体隐性或X染色体连锁隐性遗传病,其致病机制主要涉及动纤毛的结构和功能缺陷。从1999年发现DNAI1基因突变开始[6],已经有超过50个PCD致病基因相继被发现,这凸显了PCD的遗传异质性[5]。绝大部分PCD致病基因是动纤毛“9+2”微管结构的组成成分或与其运输、组装、调节相关的基因。动纤毛分布在呼吸道上皮、大脑室管膜和输卵管壁等部位。此外精子鞭毛也与动纤毛类似,它们都具有“9+2”微管结构:外侧的9组二联体微管环绕2个中央微管(central pair,CP)。中央微管与外周微管之间通过径向辐条(radial spokes,RS)连接,相邻的二联体微管通过微管连接蛋白(nexin dynein regulatory complexes,N-DRC)连接。此外,外周微管上还对接内动力臂(inner dynein arms,IDA)、外动力臂(outer dynein arms,ODA) 控制纤毛摆动频率和弯曲幅度[3-7]。
本研究涉及的2个基因均与ODA的组装相关。ODA通过动力蛋白锚定复合体(outer dynein arm docking complexes,ODA-DCs)锚定在外周微管上。ODA到达锚定位置前,需要动力蛋白轴丝组装因子(dynein axonemal assembly factors,DNAAF)招募热休克蛋白90(HSP90),辅助ODA与ODA-DCs的预组装。DNAAF1、DNAAF2和DNAAF3预组装ODA重链,DNAAF5和DNAAF6预组装ODA中链,LRRC6预组装远端纤毛ODA。
ODAD1(outer dynein arm docking complex subunit 1)基因编码外动力臂对接复合体亚基,其缺乏会导致外动力蛋白臂(ODA)无法正确组装到微管双联体。ODAD1基因全长23.62 kb,包含14个外显子,编码670个氨基酸[8]。ODAD1基因突变通常导致ODA缺失,纤毛几乎完全不动,并导致典型的PCD临床表型,包括新生儿呼吸窘迫、nNO水平低、鼻窦炎、中耳炎、支气管扩张和内脏转位等[9]。患者1的ODAD1:c.702_705dupGCAG (p.P236Afs*11)移码突变引起阅读框改变从而导致提前终止密码子产生,可能引起无义介导的mRNA降解,或产生截短蛋白质,导致蛋白质产物无法行使正常的功能,引起患者的PCD表型。
DNAAF6(dynein axonemal assembly factor 6)基因编码动力蛋白轴丝组装因子,在ODA和IDA的胞质预组装中起关键作用,这些蛋白随后通过鞭毛内运输(IFT)运输到纤毛或鞭毛轴丝。DNAAF6基因全长37.64 kb,包含7个外显子,编码214个氨基酸。DNAAF6基因突变导致ODA和IDA的缺失,影响呼吸道纤毛和精子鞭毛的运动能力[10]。DNAAF6基因突变通常导致患者出现典型的PCD表型,如慢性鼻窦炎、慢性中耳炎、慢性下呼吸道感染、支气管扩张以及nNO水平低和内脏转位等。此外,男性患者常常表现出不育的表型,精子活动能力差[11]。患者2的DNAAF6: c.532_533delCT (p.L178Sfs*2)移码突变导致提前终止密码子,可能引起无义介导的mRNA降解,导致蛋白产物无法行使正常的功能,引起患者的PCD表型。卵胞浆内单精子显微注射技术(intracytoplasmic sperm injection,ICSI)是目前PCD男性患者不育时夫妻受孕的唯一途径。有研究表明,中国目前已有携带DNAAF6变异患者通过ICSI成功生育[12],表明 ICSI 对于有生育意向的DNAAF6突变患者来说可能是一个不错的选择。
本研究在2例散发的PCD患者中分别检出已知致病基因ODAD1和DNAAF6的移码突变。2个变异均未被HGMD收录,且根据ACMG指南被判定为致病变异,支持其PCD诊断。以上结果丰富了ODAD1基因和DNAAF6基因的突变谱,有助于提高PCD的诊断率,并为PCD患者的精准治疗提供新靶点。
