反应性丙烯酸酯三嵌段共聚物与纳米SiO2协同增韧环氧树脂
2024-04-25向艳丽葛攀峰任强汪称意李坚
向艳丽, 葛攀峰, 任强 , 汪称意, 李坚
( 常州大学 材料科学与工程学院,常州 213164 )
环氧树脂具有高模量、低收缩、良好的粘结性和耐化学腐蚀性等,作为基体树脂广泛应用于复合材料[1-2]。但是因其交联固化后质脆、抗冲击性差,很大程度地限制了其在韧性要求较高材料中的应用[3-4]。环氧树脂的增韧改性通常采用热塑性弹性体[5-6]、橡胶[7-8]、核壳聚合物[9]、刚性粒子[10]等,但是往往容易出现增韧成分在环氧基体中分散不均,同时韧性和刚性难以兼顾的问题。对于弹性体等柔性材料的引入,提高环氧树脂冲击强度的同时会大幅度降低树脂体系的强度、模量及耐热性[11-12]。如何同时提高环氧树脂的韧性和刚性,仍然是一个重要的研究课题。
丙烯酸酯三嵌段共聚物由2 种或3 种丙烯酸酯类单体聚合而成的三嵌段共聚物,链段组成如ABA 或ABC 型;由于三嵌段共聚物特定结构给予了这类聚合物特定的性能,制备出的热塑性弹性体的特性被广泛应用于热固性的树脂型材的增韧改性[13-14]。刘仲良等[15]采用原子转移自由基聚合法制备了中间以聚丙烯酸丁酯为主链,两端是聚甲基丙烯酸甲酯和聚甲基丙烯酸缩水甘油酯共聚物为侧链的丙烯酸酯三嵌段共聚物(PMG-PBAPMG),将其引入到环氧树脂中对基体的韧性改善效果明显,但是其强度有一定程度降低。Redline等[16]和Caba 等[17]利用聚环氧乙烷-b-聚乙基丙烯(PEO-b-PEP)、聚环氧乙烷-b-聚氧化丙烯-b-聚环氧乙烷(PEO-b-PPO-b-PEO) 和环氧树脂共混,嵌段聚合物自组装成纳米级球形胶束,有效提高了韧性,但由于PEO、PEP、PPO 玻璃化温度较低,较高温度下共混体系表现出高应变速率敏感性,力学性能下降较多。
纳米SiO2具有高比表面积、较高的补强能力、反应活性及特殊的流变性等,广泛应用于高分子复合材料、涂料等领域[18-19]。但是纳米SiO2的表面极性强,表面能高导致其在有机相中难以分散均匀,影响其填充效果;因此有必要对其进行表面改性,改善其分散稳定性[20-21]。
课题组前期的研究表明当三嵌段弹性体聚甲基丙烯酸甲酯-b-聚丙烯酸丁酯-b-聚甲基丙烯酸甲酯(PMBM)的共聚组成为12 K-41 K-12 K 时,对于环氧树脂的增韧效果最好,但同时拉伸强度和模量相对于环氧树脂分别下降了3.5%和2.1%[22]。为了弥补这一不足,本文在保持三嵌段总分子量不变的前提条件下,分别在软段和硬段中引入含有环氧基团的甲基丙烯酸缩水甘油酯(GMA)结构单元。GMA 处于两端硬段时即PMGBMG、处于中间软段时即PMBGM,研究了GMA 结构单元在分子链上两端硬段及中间软段的不同位置对树脂增韧效果及其他性能的影响;并以硅烷偶联剂KH-550 改性的纳米SiO2接枝于该反应性嵌段聚合物上,研究嵌段聚合物与纳米SiO2协同增韧的机制及对树脂体系力学性能、热力学性能等的影响。
1 实验材料及方法
1.1 原材料
丙烯酸丁酯(BA)、甲基丙烯酸甲酯(MMA)、甲基丙烯酸缩水甘油酯(GMA)、乙酸乙酯,化学纯,上海凌峰化学试剂有限公司;溴化铜(CuBr2),化学纯,上海润捷化学试剂有限公司;五甲基二乙烯三胺(PMDETA),分析纯,阿拉丁试剂有限公司;二 α-溴代异丁酸乙二醇酯,纯度99.7%,自制;辛酸亚锡(Sn(EH)2)、甲苯、四氢呋喃(THF)、中性氧化铝、碱性氧化铝、4, 4-二氨基二苯甲烷(DDM),分析纯,国药集团化学试剂有限公司;3-氨丙基三乙氧基硅烷(KH-550):工业级,南京曙光化工有限公司;纳米二氧化硅(直径D=30 nm),99.9%,北京德科岛金科技有限公司;无水乙醇,化学纯,国药集团化学试剂有限公司;浓盐酸,37%,国药集团化学试剂有限公司;环氧树脂(E-51),工业级,南亚环氧树脂(昆山)有限公司。
1.2 反应性三嵌段丙烯酸酯共聚物的合成
参照本课题组前期工作[22-23],利用引发剂可连续再生催化剂原子转移自由基聚合(ICAR-ATRP)在75℃左右,以甲苯为溶剂合成反应性三嵌段丙烯酸酯共聚物。采用一步法合成:首先将单体BA、GMA 双官能团引发剂二α-溴代异丁酸乙二醇酯、催化剂3.5wt%CuBr2/ 无水乙醇溶液、配体PMDETA、还原剂偶氮二异丁腈(AIBN)及溶剂甲苯加入到三口圆底烧瓶中,将反应体系连续抽真空后充入氩气循环3 次,并在氩气氛围下75℃左右油浴搅拌反应6 h,使BA 和GMA 的转化率达到90% 左右,得到大分子引发剂P(BA-co-GMA),再加入MMA 单体聚合,同一温度下继续反应7 h左右,得到PMMA-b-P(BA-co-GMA)-PMMA,简写为PMBGM。如果先加入BA 单体聚合,得到PBA 大分子引发剂,然后再加入MMA 和GMA 聚合,则得到产物P(MMA-co-GMA)-b-PBA-b-P(MMA-co-GMA),简称PMGBMG。最后将得到的产物用乙酸乙酯溶解稀释后通过中性氧化铝柱子去除铜离子,减压蒸馏和甲醇沉淀去除产物中的溶剂及未反应完的单体。将最终产物铺于离型纸上晾干,80℃真空干燥48 h。
1.3 KH-550 改性纳米SiO2
将纳米SiO2、无水乙醇和蒸馏水加入到三口圆底烧瓶中;室温超声分散1 h 后,磁力搅拌的同时缓慢滴加KH-550,并用5wt% 盐酸溶液把pH 调到3 左右;在70℃磁力搅拌冷凝回流反应3 h,并用无水乙醇/H2O 混合液反复冲洗抽滤,至溶液pH=7 左右;最后将产物在冷冻干燥机(FD-1A-50,上海鑫翁科学仪器有限公司)中去除溶剂得到KH-550 改性的纳米SiO2(KH550-SiO2)。
1.4 KH550-SiO2 接枝嵌段共聚物
取适量的嵌段聚合物和乙酸乙酯于圆底三口烧瓶中,80℃磁力搅拌冷凝回流至聚合物完全溶解,然后加入相对聚合物质量1/10 的KH550-SiO2,继续搅拌回流3 h;再把接枝的嵌段共聚物用无水甲醇沉淀置于离型纸上晾干,最后50℃真空干燥24 h 即得接枝产物(嵌段共聚物-SiO2)。
1.5 共混样条的制备
1.5.1 KH550-SiO2/E-51 体系
首先取适量的环氧树脂E-51 在90℃油浴加热机械搅拌,然后加入一定量的KH550-SiO2;接着混合分散1 h 后加入固化剂DDM,抽真空排除气泡;最后倒入80℃预热的模具中固化80℃/4 h、150℃/2 h,自然冷至室温脱模。
1.5.2 嵌段共聚物-SiO2/E-51 体系
取适量的环氧树脂E-51 在120℃油浴加热机械搅拌,然后加入一定量的嵌段共聚物-SiO2混合均匀,降温至90℃加入固化剂DDM,抽真空排除气泡;最后倒入80℃预热的模具中固化80℃/4 h、150℃/2 h,自然冷至室温脱模。
1.6 测试与表征
1.6.1 凝胶渗透色谱(GPC)
采用美国Waters 公司的凝胶渗透色谱仪(GPC)测定嵌段聚合物分子量及其分布。在室温测试,四氢呋喃作为流动相,配制样品浓度0.3%~0.5%,流速1 mL/min,聚苯乙烯为标样。
1.6.2 核磁共振氢谱(1H NMR)
采用瑞士Bruker 公司的DMX-4001H NMR 对聚合物的分子结构进行表征,室温下测试,以四甲基硅烷(TMS)为内参,氘代氯仿(CDCl3)为溶剂。
1.6.3 拉伸性能测试
利用扬州天源试验机械有限公司TY-8000 型万能材料试验机,按照ASTM D638-2014[24]标准制备样条和测试强度,拉伸速度为5 mm/min。
1.6.4 断裂韧性测试
采用扬州天源试验机械有限公司TY-8000 型万能材料试验机进行三点弯曲试验,跨距为30 mm,测试速率为5 mm/min,按照国家标准GB/T 4161-2007[25]规定测定临界应力强度因子(KIC)。采用50 mm×8 mm×4 mm 的矩形样条,利用扬州天源试验机公司TY-4021 型缺口制样机在样条中部制备3 mm 左右的缺口,然后用超薄刀片在缺口根部制作一个V 形裂纹,预制裂纹与厚度之比为0.35~0.55。
1.6.5 动态力学分析(DMA)
采用美国Pekin Elmer 公司生产的DMA-8000型动力学分析仪对样条热力学性能进行分析。测试条件采用双悬臂模式、5℃/min 从-100℃升至200℃,样条尺寸为50 mm×8 mm×3 mm。
1.6.6 扫描电子显微镜及EDS 元素分析
利用德国蔡司SUPRA55 型扫描电子显微镜(SEM)对断裂表面形貌进行分析和EDS 元素分析,断面先进行喷金处理。
1.6.7 红外光谱表征
利用美国尼高力公司的Nicolet Avatar 370 型傅里叶变换红外光谱仪对样品进行溴化钾压片法红外测试,测定范围:4 000~400 cm-1,扫描16 次,分辨率为0.4 cm-1。
2 结果与讨论
2.1 嵌段共聚物数均分子量及其分布
根据课题组前期的研究,三嵌段弹性体聚甲基丙烯酸甲酯-b-聚丙烯酸丁酯-b-聚甲基丙烯酸甲酯(PMBM)的共聚组成为12 K-41 K-12 K 时对环氧树脂的增韧效果最好[22],本研究合成了两种新的含GMA 结构单元分别位于软段和硬段的三嵌段共聚物PMBGM 和PMGBMG。嵌段前后聚合物的GPC 曲线如图1 所示。可以看出嵌段前后曲线都趋于对称的单峰且嵌段后的GPC 曲线相对嵌段前明显前移,说明第一段大分子引发剂都参与了反应并成功制备了嵌段共聚物。其分子量、分子量分布(PDI)及组成见表1,可以看出嵌段共聚物相对分子量均为65×103左右,分子量分布在1.51 以下且MMA 相对总分子链的质量分数均约为40wt%。

表1 嵌段共聚物的分子量及其分布Table 1 Molecular weight and distribution of block copolymer

图1 嵌段前后的聚合物GPC 曲线Fig.1 GPC curves of polymer before and after block
2.2 嵌段共聚物的共聚组成
图2 为表1 中嵌段共聚物的1H NMR。图中a、b 分别为结构单元BA 中O-CH2亚甲基质子吸收峰和结构单元MMA 中O-CH3甲基质子吸收峰;c、d、e 分别对应于结构单元GMA 中环氧基团上的质子峰。通过峰面积之比算得PMBM-40、PMBGM 和PMGBMG 中n(BA)∶n(MMA)∶n(GMA)摩尔比分别为1.32∶1.00∶0、1.25∶1.00∶0.17、1.35∶1.00∶0.23,与实际投料比相近,进一步说明成功制备了三嵌段共聚物。

图2 嵌段共聚物的1H NMR 图谱Fig.2 1H NMR spectra of block copolymer
2.3 KH550-SiO2 及嵌段共聚物-SiO2 的官能团变化
图3 是KH-550、纳米SiO2和KH550-SiO2的红外图谱。图中3 400 cm-1左右的峰为-OH 和-NH 基团的伸缩振动峰,2 900~3 000 cm-1对应峰归属于KH-550 中的亚甲基和甲基的特征吸收峰。纳米SiO2被KH-550 改性后在2 900~3 000 cm-1出现亚甲基和甲基的特征吸收峰。在1 000~1 100 cm-1对应于Si-O-Si 基团的特征吸收峰,改性后也变得更宽、变强。说明纳米SiO2被成功改性。
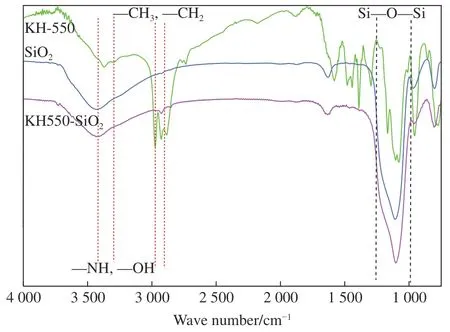
图3 KH-550、纳米SiO2 和KH550-SiO2 的红外图谱Fig.3 IR spectra of KH-550, nano-SiO2 and KH550-SiO2
图4 为嵌段共聚物-SiO2的组成及结构的红外图谱。图中在3 400 cm-1左右出现特征峰对应于-NH 和-OH 的伸缩振动峰,对比KH550-SiO2接枝的嵌段共聚物后峰明显变强,说明改性SiO2成功接枝到嵌段聚合物上;1 730 cm-1特征峰归属为羰基特征吸收峰;1 000~1 200 cm-1处归属于Si-O-Si 基团的特征吸收峰,910 cm-1为环氧基团的特征吸收峰,相较于PMGBMG 和PMBGM 中环氧吸收峰变弱,说明环氧基团与KH-550 中的氨基反应被消耗,进一步说明嵌段共聚物中成功引入了KH550-SiO2。

图4 改性的纳米SiO2 接枝嵌段共聚物前后的红外光谱Fig.4 IR spectra of block copolymers before and after grafting with modified nano-SiO2
2.4 嵌段共聚物/E-51 共混体系力学性能
先单独进行了纳米SiO2对于环氧树脂的性能影响研究。图5 为不同SiO2添加量环氧树脂体系的临界应力强度因子KIC及拉伸性能,可以看出当纳米SiO2添加量为0.5wt%时,树脂体系KIC达到最大值2.15 MPa·m1/2,相对纯E51 提高了38%,但增加幅度有限,同时拉伸强度和弹性模量也达到了最大值。说明纳米SiO2不但可以提高树脂韧性而且对树脂的拉伸性能也有一定的改善作用,归因于其表面丰富的活性基团。但随着添加量的增加,韧性和拉伸性能变差,是由于纳米SiO2团聚形成缺陷导致树脂体系性能下降[21]。
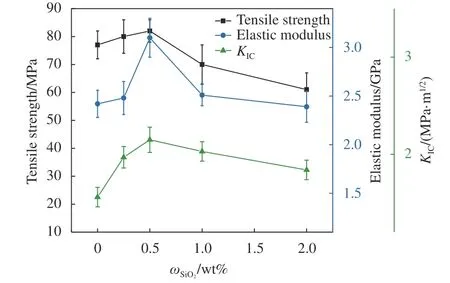
图5 不同SiO2 添加量环氧树脂体系临界应力强度因子KIC 及拉伸性能Fig.5 Critical stress intensity factor KIC and tensile properties of epoxy resin system with different amounts of SiO2
表2 为5wt% 添加量的嵌段共聚物-SiO2增韧环氧树脂的力学性能。可以看出GMA 处于两端硬段的PMGBMG 相对GMA 处于中间软段的PMBGM 对E51 的增韧效果较好。GMA 的引入主要是为了增加三嵌段聚合物形成的分散相和环氧树脂连续相的相互作用,但同时也可能会限制三嵌段聚合物软段微区的可变形能力,从而降低增韧效果。从结果来看,也印证了这一假设。中间软段PBA 相含有GMA 的嵌段共聚物由于PBA 相与环氧树脂的共交联,对于软段可变形能力的限制更多,从而导致其增韧环氧树脂的效果下降较明显。GMA 位于硬段的PMGBMG 增韧效果和原来不含GMA 的PMBM-40 效果相当。

表2 嵌段共聚物-SiO2 增韧环氧树脂的拉伸性能和断裂韧性Table 2 Tensile properties and fracture toughness of block copolymer-SiO2 toughened epoxy resin
加入纳米SiO2后树脂体系增韧效果都有明显的提高,一方面是由于纳米SiO2与环氧树脂之间易形成强相互作用力,当断裂时可消耗大量的能量;另一方面由于刚性粒子可以阻碍微裂纹和局部应力的集中导致的屈服可吸收大量能量。其中加入5%PMGBMG-SiO2嵌段共聚物增韧效果最好,相对于纯环氧树脂其KIC提高了145%,拉伸强度和模量分别提高了8%和31%,实现了韧性和刚性的同时提升。体系中引入SiO2粒子后,在应力作用下SiO2可起到应力集中作用,当SiO2与硬段链接时,应力能迅速通过硬段向PBA 软段传递,PBA 通过链段运动耗散大量能量,这种界面应力传递方式充分发挥了SiO2和PBA 软段的协同增韧效果。然而,PMBGM-SiO2粒子中PBA 软段与较硬的SiO2连接,这种结构反而降低了PBA 软段微区的变形能力,削弱了粒子的增韧效果。
2.5 嵌段共聚物/E-51 共混体系断面形貌
图6 为嵌段共聚物增韧树脂体系及纯E-51 的断面形貌。纯E-51 呈典型的脆性断裂行为,断面主要由不同高度的较光滑平面构成且几乎观察不到明显的塑性形变。与纯E-51 相比,添加嵌段共聚物后材料的断面粗糙度均出现不同程度的增大,其中PMBGM 增韧体系的断面粗糙程度增加幅度最小,这与力学性能分析结果一致;PMGBMG和PMBM-40 粗糙度相当,但PMBM-40 与基体环氧树脂之间未发生共交联反应,嵌段聚合物的自组装行为使PBA 相尺寸较大,从而形成的孔洞较大且均匀性较差;而PMGBMG 由于硬段与环氧树脂共交联反应的发生,使相尺寸减小,体系中孔洞较小且较均匀。这与He 等[26]和Kong 等[27]在采用含GMA 的丙烯酸酯聚合物增韧环氧树脂时发现的结果一致。这种较小的分散相尺寸也有利于纳米SiO2分散在三嵌段聚合物形成的分散相与环氧树脂连续相的界面上,会提高界面层材料的模量。根据复合材料的界面层拘束理论,当界面材料的模量处于分散相和连续相之间时,可以更好的传递应力,从而进一步提升PMGBMG 接枝SiO2对环氧树脂增韧效果[22,28]。

图6 5wt%添加量的嵌段共聚物增韧E-51 体系的断面形貌:((a), (b)) E-51;((c), (d)) 5%PMBM-40/E-51;((e), (f)) 5%PMBGM/E-51;((g), (h)) 5%PMGBMG/E-51Fig.6 Section morphologies of 5wt% addition block copolymers toughened E-51 system: ((a), (b)) E-51; ((c), (d)) 5%PMBM-40/E-51;((e), (f)) 5%PMBGM/E-51; ((g), (h)) 5%PMGBMG/E-51
图7 为PMGBMG-SiO2/E-51 体系的断面形貌。由图7(a)可以看出此体系下树脂断面粗糙度明显进一步提高,且裂纹相互交错,说明该体系具有较好的抗断裂韧性;从图7(b)可以看出在断裂面上均匀分布着纳米粒子及孔洞,从EDS 图谱(图7(c))可以看出硅元素总体较均匀分布在断面上,但同时有部分区域比较集中,其质量含量约为0.12wt%,折算成SiO2含量约为0.26wt%,比理论含量0.5wt%低,这是由于有部分SiO2被聚合物包裹不能被检测到。部分比较集中的区域可能是三嵌段聚合物分散相与环氧树脂连续相的界面。

图7 5%PMGBMG-SiO2/E-51 混合体系断面形貌((a), (b))及EDS 测试硅元素分布图(c)Fig.7 Cross section morphologies of 5%PMGBMG-SiO2/E-51 thermosets ((a), (b)) and silicon distribution map from EDS (c)
2.6 嵌段共聚物/E-51 共混物的热机械性能
图8 为嵌段聚合物/E-51 体系的DMA 图谱。可以看出在低温部分的玻璃化转变温度Tg在-50℃左右,主要是双酚A 型环氧树脂在较低温度下的二级转变(β 转变)[29]。对于储存模量,在50℃之前,纯E-51 和共混物的模量相当,但是当温度超过75℃,5%PMGBMG-SiO2/E-51 比5%PMGBMG/E-51 和E-51 可以更好地保持模量,直到进入玻璃化转变区才开始下降。从损耗模量曲线来看,5%PMGBMG/E-51、5%PMGBMG-SiO2/E-51 和纯E-51 的Tg分别为158℃、166℃和173℃,嵌段共聚物的加入使树脂体系的耐热程度有小幅下降,但5%PMGBMG-SiO2/E-51 体系的耐热性相对于5%PMGBMG/E-51 保持较好;主要是由于嵌段共聚物作为弹性体增韧环氧树脂,其本身耐热性低于环氧树脂,而SiO2有较高的刚性和补强作用,会相对提升混合树脂体系的耐热性。说明PMGBMGSiO2协同增韧体系不但具有较好的增韧效果还可以较好地保持树脂体系的耐热性。

图8 环氧树脂及增韧环氧体系DMA 曲线Fig.8 DMA curves of epoxy and toughening epoxy thermosets
3 结 论
(1) 利用引发剂可连续再生催化剂原子转移自由基聚合(ICAR-ATRP) 合成了不同结构的反应性嵌段聚合物聚甲基丙烯酸甲酯-b-聚(丙烯酸丁酯-co-甲基丙烯酸缩水甘油酯)-b-聚甲基丙烯酸甲酯(PMBGM)和聚(甲基丙烯酸甲酯-co-甲基丙烯酸缩水甘油酯)-b-聚丙烯酸丁酯-b-聚(甲基丙烯酸甲酯-co-甲基丙烯酸缩水甘油酯) (PMGBMG),并用硅烷偶联剂KH-550 改性的纳米SiO2成功接枝于该嵌段聚合物。
(2) 甲基丙烯酸缩水甘油酯(GMA)处于中间软段的PMBGM 增韧环氧树脂其刚性性能降低较多,韧性略有提升;GMA 处于两端硬段的PMGBMG的增韧环氧树脂相对纯环氧树脂E-51 其刚性性能基本相同,韧性大幅提升且与不含有GMA 的聚甲基丙烯酸甲酯-b-聚丙烯酸丁酯-b-聚甲基丙烯酸甲酯(PMBM-40)效果相当。
(3) 纳米SiO2与GMA 处于两端硬段的嵌段共聚物PMGBMG 协同增韧环氧树脂效果最佳,相对纯环氧树脂临界应力强度因子(KIC)提高了145%,拉伸强度和模量分别提高了8%和31%,起到了韧性和刚性同时提升的效果,且耐热性保持较好。该研究为环氧树脂同时增韧和增强提供了一种新的方案。
