基于权重基因共表达网络分析挖掘人参皂苷Rh1生物合成相关基因
2024-04-22刘思章于靖辉张美萍
刘 畅,刘思章,于靖辉,陈 屏,雷 军,王 义,张美萍
• 药材与资源•
基于权重基因共表达网络分析挖掘人参皂苷Rh1生物合成相关基因
刘 畅,刘思章,于靖辉,陈 屏,雷 军,王 义,张美萍*
吉林农业大学生命科学学院,吉林省人参基因资源开发与利用工程研究中心,吉林 长春 130118
通过权重基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)挖掘人参皂苷Rh1生物合成相关基因。以种植于吉林省人参主产区304个农家品种的转录组测序数据为基础,以人参皂苷Rh1含量为表型进行差异表达基因的挖掘。通过WGCNA获得6个与人参皂苷Rh1含量密切相关的基因共表达模块,根据关联分析结果确定Green模块为与人参皂苷Rh1含量显著相关的关键模块。功能注释结果显示Green模块内的基因富集到mRNA结合、蛋白修饰等多个相关途径。通过构建基因互作网络筛选获得7个核心基因,功能预测表明这些基因可能在人参皂苷Rh1的生物合成途径中起着重要作用。对7个候选基因进行茉莉酸甲酯(methyl jasmonate,MeJA)体外调控分析。对人参不定根进行MeJA诱导处理,随MeJA的诱导,、、、的表达量与人参皂苷Rh1的含量同时呈现显著变化,但只有的表达趋势与人参皂苷Rh1在MeJA诱导下的表达趋势相同,表明这条候选基因可能与人参皂苷Rh1的合成密切相关。为人参皂苷Rh1生物合成途径的解析提供了理论基础。
人参;人参皂苷Rh1;差异表达基因;权重基因共表达网络分析;茉莉酸甲酯
人参C. A. Meyer是一种药用价值极高的五加科人参属多年生草本植物,广泛应用于中药领域。其根、茎、叶和花等部位都含有丰富的生物活性成分,如人参皂苷、多糖、维生素、氨基酸等。人参皂苷作为最重要的活性成分之一,具有提高免疫力、抗氧化、改善中枢神经系统紊乱等多种药理作用[1]。目前已从人参中分离鉴定出多种不同的人参皂苷,如人参皂苷Rh1、Rc、Rd、Rf等,这些不同种类的人参皂苷具有不同的药理活性和生物利用度。随着生物信息学技术的发展,利用基因组学、转录组学、代谢组学等手段对人参遗传信息进行解析已经成为人参皂苷合成途径研究的重要手段。这些研究不仅揭示了人参皂苷合成途径的分子机制,还为人参的遗传育种提供了重要的理论基础。
人参皂苷Rh1是一种四环三萜达玛烷型皂苷单体,按结构分类属于原三醇型人参皂苷,是一种具有广泛药理活性的稀有皂苷[2-3]。已有研究表明,人参皂苷Rh1可以抑制肿瘤细胞的扩散,对神经系统和免疫系统也有积极的影响,具有改善哮喘[4]、抗癌[5]、预防肝损伤[6]多种药理作用。人参皂苷Rh1的生物合成途径包括3个部分:上游人参皂苷母核苷元前体物的合成、中游人参皂苷母核苷元碳骨架延长以及下游母核苷元形成及后修饰。其中,上游人参皂苷母核苷元前体物的合成是人参皂苷Rh1合成途径中的关键步骤之一。这些前体物质可以通过甲羟戊酸(mevalonic acid,MVA)和甲基赤藓糖醇磷酸化(methylerythritol phosphate pathway,MEP)途径合成,其中异戊二烯焦磷酸(isopentenyl pyrophosphate,IPP)和二甲基烯丙基焦磷酸(dimethylallyl pyrophosphate,DMAPP)是合成人参皂苷母核苷元骨架的重要前体物。中游人参皂苷母核苷元碳骨架延长是人参皂苷Rh1合成途径的第二个关键步骤。IPP和DMAPP在多种萜类合酶的催化下,依次形成牻牛儿基焦磷酸(geranyl pyrophosphate)、法尼基焦磷酸(farnesyl pyrophosphate)和角鲨烯(squalene),完成了人参皂苷母核苷元骨架的延长。角鲨烯通过角鲨烯环氧酶形成不同人参皂苷母核苷元的共同前体物质2,3-氧化角鲨烯,为人参皂苷Rh1的合成提供了重要的前体物质。下游母核苷元形成及后修饰是人参皂苷Rh1合成途径的最后一个步骤。这一步骤主要包括一系列的细胞色素氧化酶(P450)、糖基转移酶等酶的修饰。通过这些酶的作用,人参皂苷Rh1的结构得到了进一步的修饰和调整,最终形成了人参皂苷Rh1单体[7-9]。
转录组测序是目前广泛应用于生命科学研究中的一种高通量技术。它可以帮助研究人员准确鉴定和获得性状功能基因。然而,由于基因的表达与性状间存在复杂的关联,因此我们需要发现一种更为精准的方法来挖掘控制目标性状的候选基因[10-11]。差异表达基因(differentially expressed genes,DEGs)是指当基因在RNA水平处于不同环境、压力、和时间等条件时,表达有显著性差异的基因[12]。近年来差异表达基因相关分析在药用植物研究领域中广泛应用,对控制相关生物学基因性状的候选基因鉴定以及植物品种选育具有重要意义[13-17]。权重基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)作为一种新的基因互作网络建模方法,能够将基因分为不同的模块,并与表型相关联,从而挖掘出关键模块中的基因进行深入研究[18-19]。WGCNA已经被广泛应用于药用植物领域[20-22]。然而,在人参研究领域中,尚未有研究使用WGCNA的分析方法挖掘与人参皂苷生物合成相关的基因。人参皂苷是人参中最主要的有效成分之一,对于揭示其生物合成机制具有重要意义。因此,使用WGCNA方法,挖掘与人参皂苷合成相关的基因,将有助于深入了解人参皂苷的生物合成机制,提高人参的遗传育种水平。
本研究以吉林省人参主产区304个农家品种的转录组测序数据为基础,并挖掘了与人参皂苷Rh1差异表达相关的基因,构建了WGCNA共表达网络。通过将共表达网络中的基因划分为不同的基因模块,再通过与人参皂苷Rh1含量的相关性分析进一步确定关键模块。筛选出人参皂苷Rh1的生物合成相关中的核心基因,该核心基因在人参皂苷Rh1的生物合成途径中具有重要作用,本研究还对这些核心基因的表达模式进行了分析,为深入了解人参皂苷Rh1的生物合成途径提供了参考。同时为实现利用植物细胞工厂定向化生产人参皂苷Rh1奠定了基础。为其他药用植物重要性状相关基因挖掘提供参考。
1 仪器与材料
1.1 仪器
Waters 2695型高效液相色谱仪,美国Waters公司;电子分析天平,上海精密科学仪器有限公司;超声清洗机,宁波新芝生物科技有限公司;37 ℃恒温烘箱,Thermo公司;恒温摇床,上海智诚分析仪器制造有限公司;超低温冰箱,Thermo公司;水浴锅,上海一恒科技有限公司;旋转蒸发器,上海亚荣生化仪器厂;荧光定量PCR仪,赛默飞世尔科技有限公司。
1.2 材料
对照品人参皂苷Rh1(批号为B21061),质量分数≥98%,上海源叶生物科技有限公司;甲醇(批号M813895)购自上海麦克林生化科技股份有限公司;色谱甲醇批号为MS1922-801,色谱乙腈批号为AS1122-801,美国天地有限公司;屈臣氏蒸馏水,北京屈臣氏蒸馏水有限公司;茉莉酸甲酯(methyl jasmonate,MeJA)批号为M46630,上海吉至生化科技有限公司。
本研究所用304份人参材料为吉林省人参基因资源开发与利用工程研究中心提供,来源于种植在吉林省人参主产区的四年生人参主根,经吉林农业大学生命科学学院王义教授鉴定为五加科人参属植物人参C. A. Meyer;本研究所用数据库为吉林省人参基因资源开发与利用工程研究中心根据种植于吉林省人参主产区的304个农家品种(S1~S304)4年生人参样本根部转录组测序数据所建立的转录组序列及表达量数据库(NCBI: SRR1313640~SRR132505)[23],包括304个农家品种4年生人参样本根部人参皂苷Rh1含量。
2 方法
2.1 人参皂苷Rh1生物合成差异基因的挖掘
2.1.1 样本数据的预处理 将数据库中的304个样本按照人参皂苷Rh1含量进行排序,分别取人参皂苷Rh1含量较高的前25个样本及人参皂苷Rh1含量较低的后25个样本,设置为高含量组及低含量组,绘制布鲁克斯图,探讨样本间差异性。使用Perl语言分别调取2组样本的转录组序列及表达量,用于差异基因挖掘。
2.1.2 差异基因的挖掘 调用R语言中的DESeq2包对上述高低2组样本中的数据样本进行差异基因分析,将≤0.05的基因视为差异基因。利用TBtools绘制差异基因的火山图及热图,并通过主成分分析(principal component analysis,PCA)进一步分析验证高低两组样本间的差异性。
2.2 权重共表达网络的构建
利用权重共表达网络分析基因与人参皂苷Rh1含量之间的关系,以“2.1.2”项中挖掘获得的差异基因为输入数据,调用R语言中的WGCNA包构建共表达网络。利用Pearson相关矩阵和网络拓扑分析分别计算基因相关性和软阈值能力。选择合适的软阈值对原始矩阵进行转化,得到邻接矩阵,然后将邻接矩阵转化为拓扑重叠矩阵,并运用动态切割法进行基因聚类及划分模块。设定模块最少基因数为5,相似模块合并阈值为0.85。绘制样本表达模式热图,并通过模块特征值来展示模块基因在各个样本中的表达模式,根据模块特征向量基因分析确定与人参皂苷Rh1含量显著相关的特异性模块,选择相应的模块进行深入研究。
2.3 关键模块的功能富集分析
为进一步分析关键模块的功能,使用Blast2GO及京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)在线平台对关键模块内的基因进行基因本体论(GeneOntology,GO)功能注释及KEGG通路分析。经过多重检验校正后,以≤0.05为阈值,满足此条件的定义为显著富集的GO条目及KEGG 通路。
2.4 基因互作网络构建及核心基因的挖掘
为获得关键模块中的核心基因,为获得关键模块中的核心基因,利用Cytoscape3.9.0软件中的cytoHubba插件,选择MCC算法,对关键模块内的基因进行可视化处理,每个节点代表1个基因,连线代表基因间的关系。根据互作网络快速筛选出Weight值及模块连通性都较高的基因,作为关键模块核心基因。利用NCBI数据库(https://www. ncbi.nlm.nih.gov/)获取这些核心基因的相关信息,并在韩国人参基因组[24]中查找其同源基因的注释结果,进一步预测其功能。
2.5 MeJA诱导人参皂苷Rh1
将2 g在固体B5培养基上生长的人参不定根接种于液体B5培养基中,在22 ℃、110 r/min的摇床中黑暗培养23 d,在对数生长期后,向培养基中添加200 μmol/L MeJA进行诱导处理,诱导时间设置为6、12、24、36、48、60、72、84、96、108、120 h,在每个时间点收获材料,同时以MeJA处理0 h的材料作为对照组收获,对上述每组材料进行3次生物学重复。取样时,将0.5 g新鲜样品进行液氮速冻,保存在−80 ℃冰箱中,将剩余样品在37 ℃恒温烘箱烘干至恒定质量后提取人参皂苷。利用索氏提取法进行皂苷提取。
2.6 人参皂苷Rh1的测定
2.6.1 对照品溶液的制备 分别称取人参皂苷Rh1对照品适量,加入甲醇定容,经0.22 µm膜滤过,制成含人参皂苷Rh1质量浓度为0.075 mg/mL的对照品溶液。
2.6.2 供试品溶液的制备 取“2.5”项所述方法的人参皂苷提取液,用0.22 µm有机滤膜滤过,得供试品溶液。
2.6.3 色谱条件[25]高效液相色谱系统是Waterse2695,色谱柱为Waters C18柱。流动相组为屈臣氏蒸馏水(A)和色谱乙腈(B),梯度洗脱,进样量为10 µL,柱温30 ℃,流动相体积流量为1.0 mL/min。检测波长为203 nm。梯度洗脱:0~40 min,18%~21% B;40~42 min,21%~26% B;42~46 min,26%~32% B;46~66 min,32%~33.5% B;66~71 min,33.5%~38% B;71~86 min,38%~65% B;86~91 min,65% B;91~96 min,65%~85% B;96~103 min,85% B;103~105 min,85%~18% B;105~120 min,82% B。
2.6.4 线性关系考察 精密吸取上述混合对照品溶液,按照“2.6.3”项色谱条件进行测定,以色谱峰峰面积为纵坐标(),以人参皂苷Rh1对照品质量浓度为横坐标(),绘制标准曲线,进行线性回归,得到回归方程,相关系数(2)和线性范围分别为人参皂苷Rh1=347 766.69-6 332.14,2=0.999 65,线性范围6.0~0.375 μg/mL。
2.6.5 稳定性试验 精密吸取适量的人参皂苷Rh110 μL对照品溶液,依照“2.6.3”项色谱条件分别在0、4、8、12、16、20、24 h进样,测得人参皂苷Rh1的峰面积的RSD为0.80%,表明仪器稳定性良好。
2.6.6 精密度试验 精密吸取适量的人参皂苷Rh110 μL对照品溶液,按照“2.6.3”项色谱条件进行分析,连续进样6次,测得人参皂苷Rh1的峰面积的RSD为0.89%,表明仪器精密度良好。
2.6.7 重复性试验 精密吸取人参皂苷Rh1对照品溶液6份,按照按照“2.6.3”项色谱条件进行分析,测定人参皂苷Rh1的峰面积,通过线性回归方程计算其质量分数RSD值为0.96%,结果表明该方法重复性较好。
2.6.8 加样回收率的测定 精密吸取已知质量分数的人参皂苷Rh1对照品溶液,按照“2.6.2”项和“2.6.3”项方法制备供试液和进样,计算得到人参皂苷Rh1的加样回收率为99.63%,RSD值为1.14%,数据表明测试结果的准确度良好。
2.6.9 人参皂苷Rh1的测定 根据“2.6.3”项色谱条件对MeJA处理的人参不定根中的人参皂苷Rh1进行含量测定。
2.7 RNA提取、cDNA文库构建及数据组装
使用Trizol试剂盒提取人参不定根的总RNA,按照Jiang等[26]的方法构建RNA-seq文库。文库经过鉴定、定量和复用,使用HiSeq X Ten(Illumina,Inc.,San Diego,美国)进行测序,使用Trinity (Version 2.14.0)软件,以人参unigene的转录组为参考,从过滤后的读数中组装单个基因转录本。使用RSEM(Version 1.3.3)软件对单个转录本的表达量和基因的整体表达量进行了量化。转录本的表达量以每百万转录本(TPM)为单位,用于进一步分析。PgRh候选基因转录本的表达量是从每个时间点取样的不定根的每个生物重复中提取的。
2.8 人参皂苷合成候选基因在MeJA诱导下的功能验证
为了验证MeJA处理对人参皂苷Rh1生物合成的影响,通过检验,将6~120 h每个时间点的3个生物重复的人参皂苷Rh1含量与0 h时间点的人参皂苷Rh1含量进行比较。使用TRIpure Reagent Total RNA Extraction Reagent从每个时间点采样的3个生物重复中分离出总RNAs。PgRh候选基因转录本的表达量是从每个时间点取样的不定根的每个生物重复中提取的。通过检验,比较每个时间点MeJA处理的样本和0 h时间点对照样本的基因表达,确认MeJA处理对PgRh候选基因表达的影响。为了进一步确认PgRh候选基因在人参皂苷Rh1生物合成中的作用,分析其单个转录本的表达,并对每个时间点(包括0 h)的单个转录本的表达与每个时间点的人参皂苷Rh1含量进行了Pearson相关分析。如果一个PgRh候选基因的转录本表达量与人参皂苷Rh1含量在≤0.05的双尾显著性水平下有相关性,则认为拼接到该转录本的基因参与人参皂苷Rh1的生物合成。
3 结果与分析
3.1 人参皂苷Rh1生物合成差异基因的挖掘
3.1.1 样本数据的预处理 首先对304个农家品种4年生根的人参皂苷Rh1含量分布情况进行了统计。如图1所示,304个农家品种4年生根中人参皂苷Rh1皂苷含量呈正态分布,存在较大差异。将304个农家品种4年生根中的人参皂苷Rh1含量进行分组,每组25个农家品种,含量高的为高组,含量低的为低组。图2显示了这2组人参皂苷Rh1皂苷含量显著不同(≤0.05),可用于进一步进行差异表达基因分析。
3.1.2 差异基因的挖掘 将“3.1.1”项得到的差异基因进行了火山图(图3)和热图的绘制(图4)。在703个差异基因中,有578个基因上调,而125个基因下调。对这些差异基因进一步进行了PCA分析。PCA结果(图5)表明高低含量2组之间存在明显差异,这为进一步的分析提供了基础。
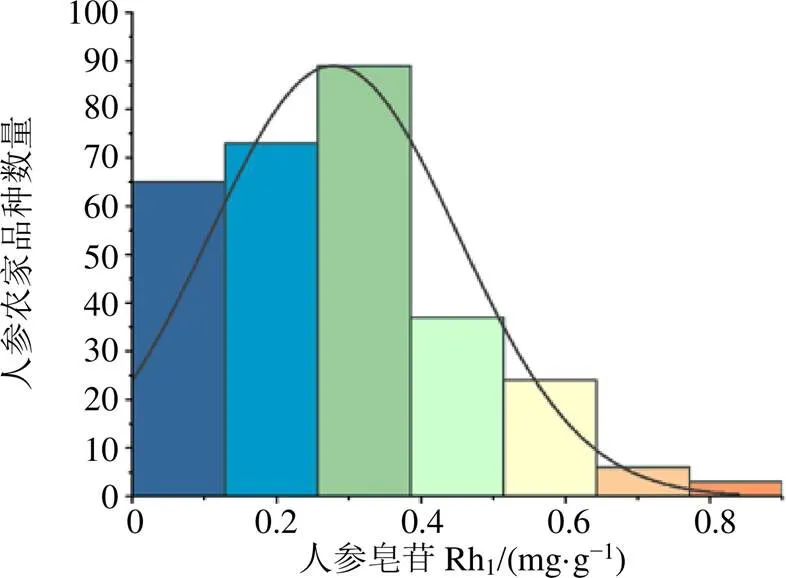
图1 304份样本人参皂苷Rh1含量的频率分布直方图
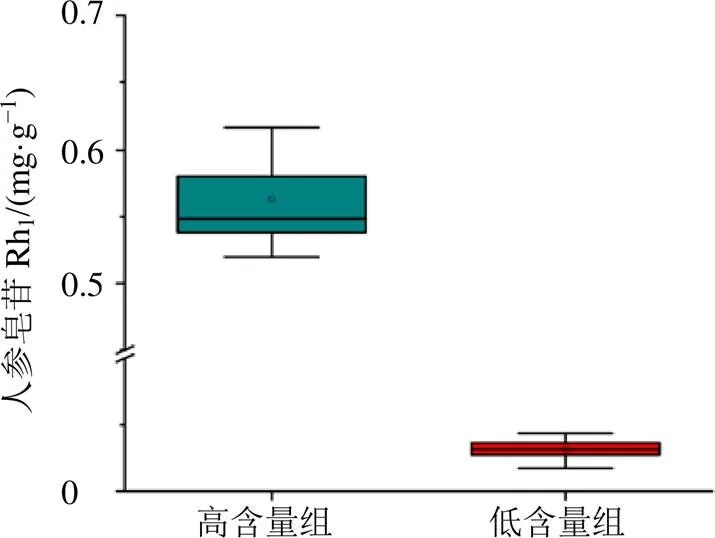
图2 人参皂苷Rh1含量差异分组
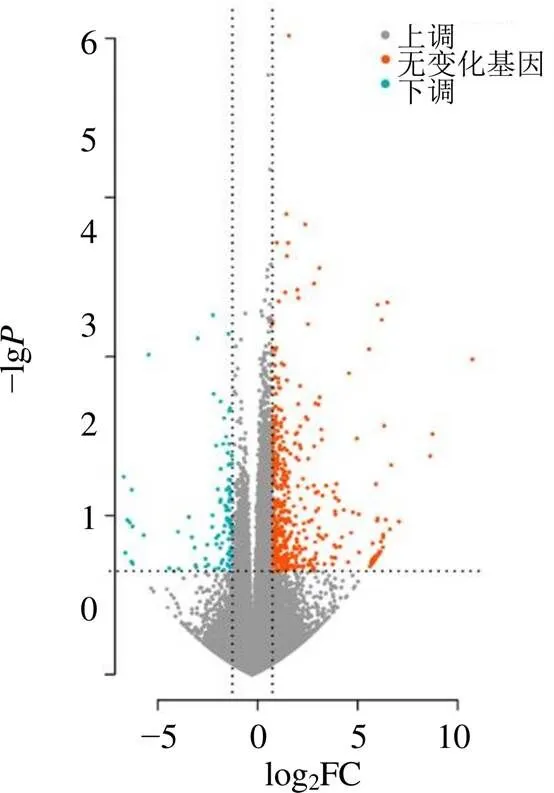
图3 人参皂苷Rh1差异基因火山图
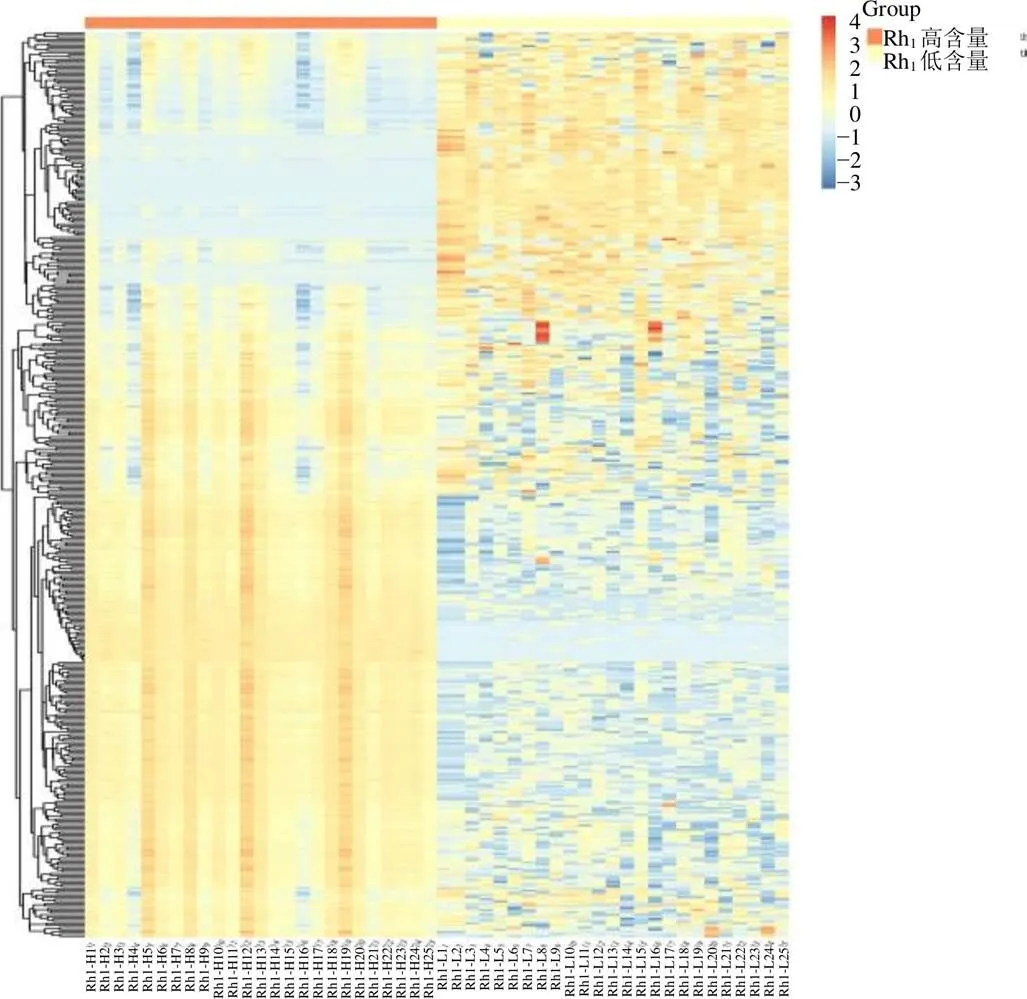
图4 人参皂苷Rh1差异基因表达量热图
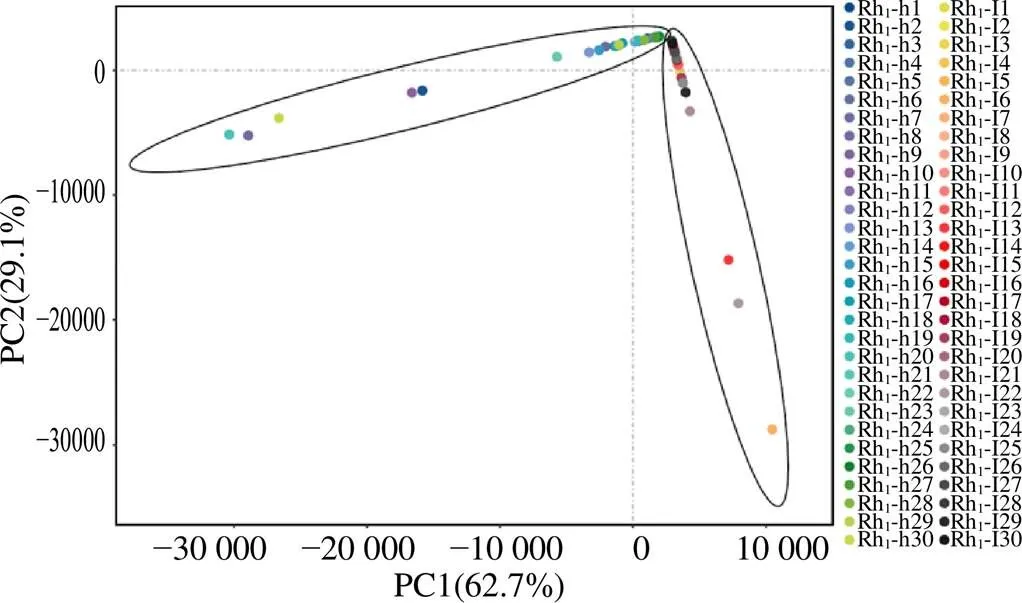
图5 人参皂苷Rh1差异基因PCA分析
3.1.3 差异基因的功能富集分析 对“3.1.2”项获得的差异基因进行了GO功能注释和KEGG信号通路分析。进一步总结了差异基因的GO功能注释和KEGG信号通路分析结果。根据GO功能注释的结果(图6),发现核心模块基因的功能主要涉及核酸结合(GO: 0003676)、RNA结合(GO: 0003723)、mRNA结合(GO: 0003729)、细胞液(GO: 0005829)、核质(GO: 0005654)、染色质组织(GO: 0006325)、细胞蛋白修饰过程(GO: 0006464)、质体(GO: 0009536)、转移酶活性(GO: 0016740)、蛋白修饰过程(GO: 0036211)和催化活性(GO: 0140096)等多个GO term。KEGG富集结果显示(图7),差异基因的代谢通路主要集中在碳水化合物代谢、转运、信号转导、运输和分解代谢等途径。
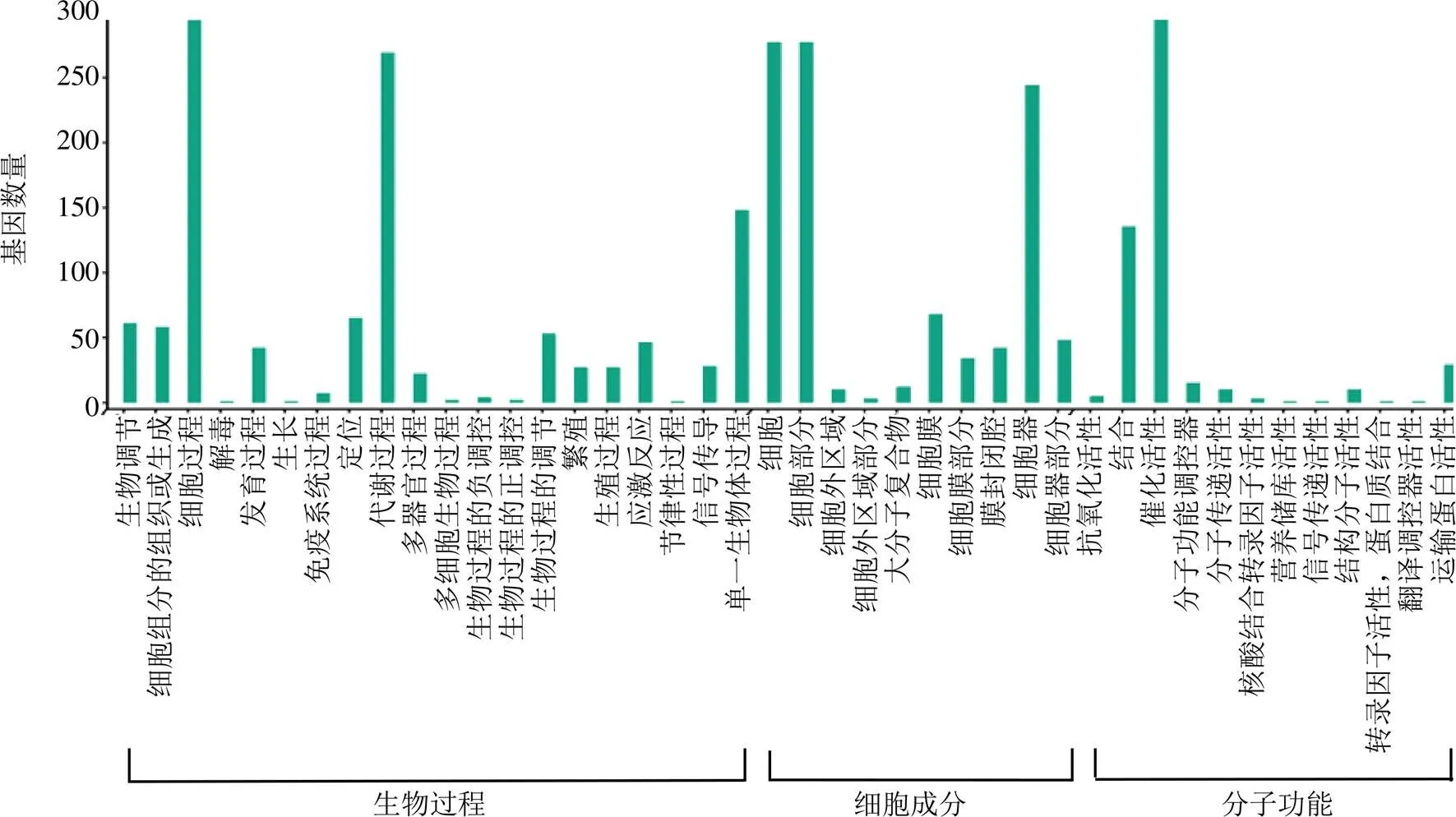
图6 核心模块的GO富集分析
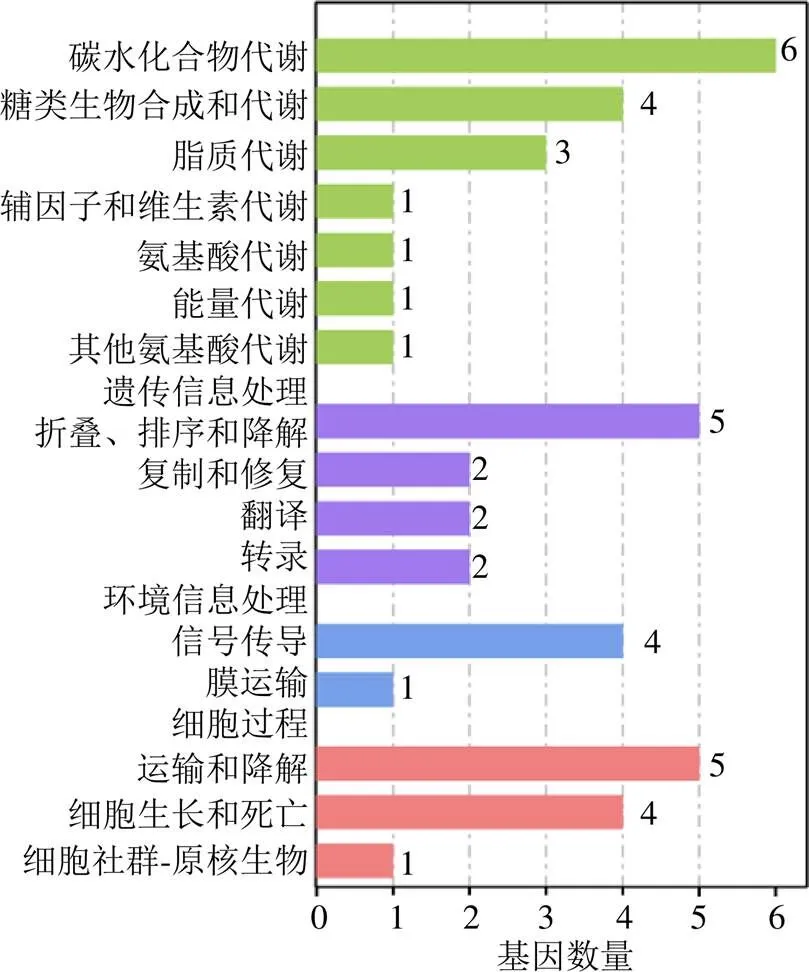
图7 核心模块KEGG富集功能分析
3.2 权重共表达网络的构建
3.2.1 软阈值的确定 根据加权共表达网络的要求,利用无尺度拓扑结构准则来确定软阈值β。运用R语言中WGCNA包中的pickSoftThreshold函数计算软阈值。数值范围设置为1~20,计算相关系数的平方和基因的平均连接度,以确定最佳的软阈值。根据本研究的选择标准,选择拟合曲线第一次超过0.85时的power值来筛选共表达模块,当power=12时,无标度拓扑拟合指数曲线在达到较高值后开始变平(图8)。因此,选择12作为后续分析的power值。
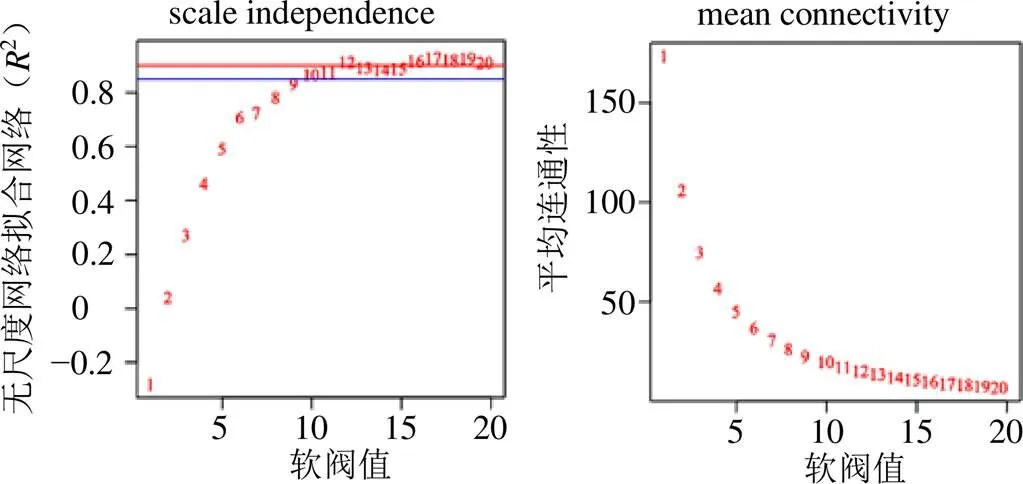
图8 软阈值的确定与平均连通度图
3.2.2 构建加权共表达网络和基因模块的确定 在网络构建和模块划分的过程中,选择了β值为12为最佳软阈值,并根据基因拓扑重叠矩阵的相异度进行聚类。通过聚类分析,得到了基因拓扑重叠聚类树和相关性热图。根据图9的结果,将与人参皂苷Rh1相关的703个差异基因分为了6个模块。在这6个模块中,turquoise模块拥有最多的基因数量,共有333条基因,而red模块则只包含6条基因,是基因数量最少的模块。一般WGCNA中grey模块被视为不能分配给任何其他模块的基因集合。
3.2.3 模块与人参皂苷Rh1的相关性系数计算及关键模块确定 通过计算基因模块与人参皂苷Rh1的相关性系数,确定了关键模块。根据图10的相关性热图,发现在6个基因模块中,有4个基因模块与人参皂苷Rh1呈现正相关,而另外2个基因模块与人参皂苷Rh1呈现负相关。在这些基因模块中,Green模块的相关系数达到了0.74,值为0.008。因此,选择Green模块中的基因进行研究。
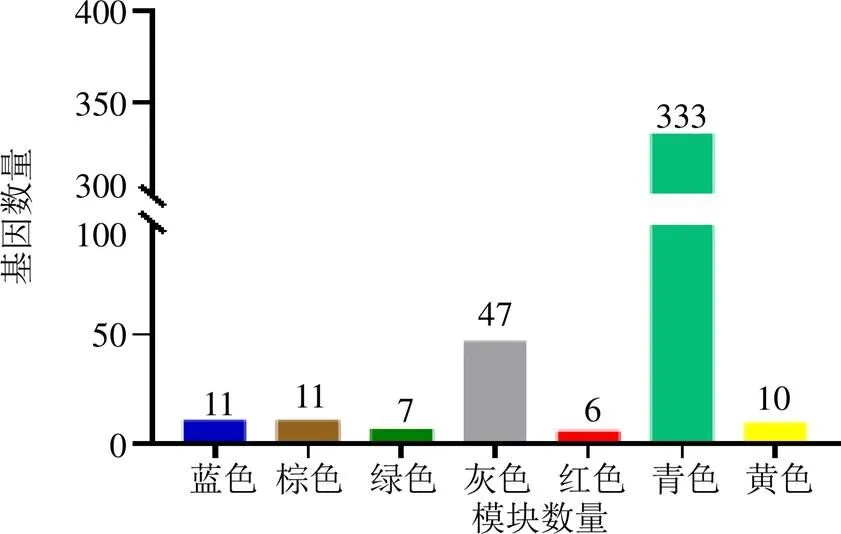
图9 6个基因模块中基因数量分布
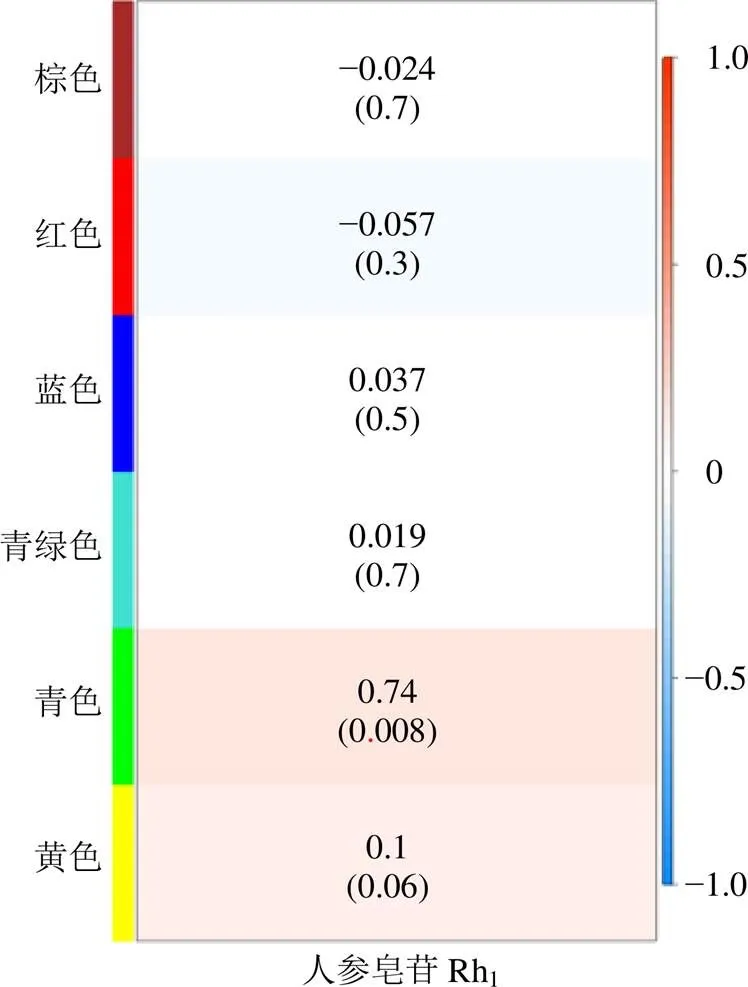
图10 6个基因模块与人参皂苷Rh1含量关联热图
3.2.4 基因互作网络与核心基因挖掘 可视化得到的核心基因进一步在NCBI数据库与韩国基因组数据库进行比对(表1)。用Cytoscape软件对基因互作网络进行可视化,根据weight值与基因之间的连通性建立基因互作网络(图11)。
3.3 MeJA诱导处理后人参皂苷Rh1含量变化
本实验在MeJA诱导处理后的不同时间内,通过高效液相色谱检测人参不定根中人参皂苷Rh1皂苷含量。根据图12结果,与对照组(0 h)相比,MeJA处理48 h后人参皂苷Rh1含量显著变化,表明人参不定根中的人参皂苷Rh1对MeJA有响应作用。MeJA显著促进了人参皂苷Rh1的生物合成,随着MeJA处理时间的增加,人参皂苷Rh1含量显著增加。在处理84 h时,人参皂苷Rh1含量达到最高值1.09 mg/g,相较于对照组,人参皂苷Rh1含量增加了3.19倍,说明MeJA对人参不定根中人参皂苷Rh1的合成起到了促进作用。
在MeJA处理的不同时间点(48、72、84、96、108、120 h)下,人参皂苷Rh1的含量与对照组相比均发生了变化。人参皂苷Rh1在MeJA处理下的48 h时与对照组相比发生显著差异,在84 h时人参皂苷Rh1含量达到顶峰。由图13可知,在MeJA处理72 h时,人参皂苷Rh1候选基因、和的表达量与对照组也发生了显著变化。同样地,在MeJA处理108 h时,人参皂苷Rh1候选基因的表达量与对照组也发生了显著变化。但是在MeJA处理48 h和84 h时,、和的表达情况与人参皂苷Rh1含量没有相同的趋势,以上结果说明只有候选基因在人参皂苷Rh1合成过程中可能发挥重要作用。

表1 Green模块中核心基因的功能注释
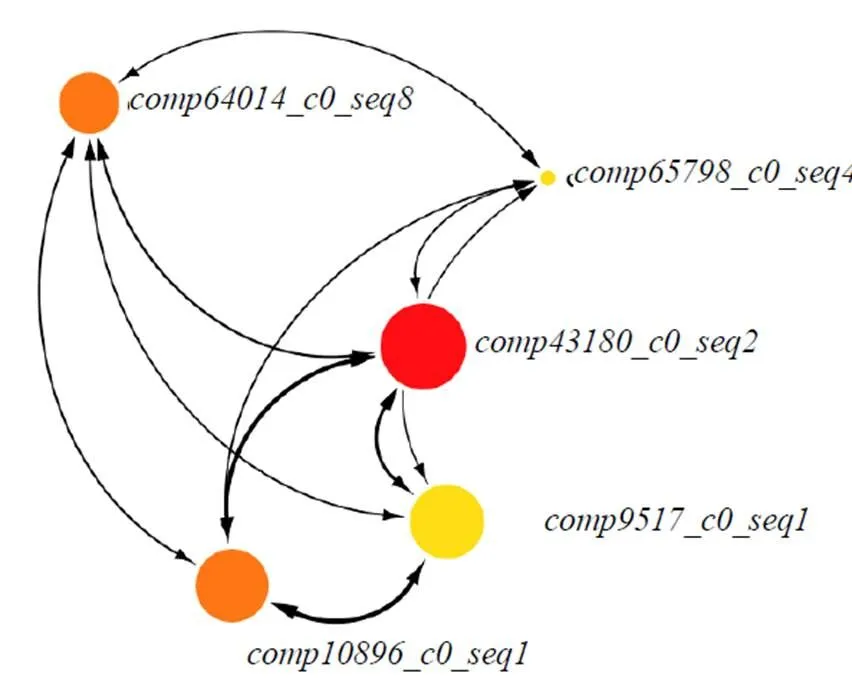
图11 核心模块中核心基因互作网络分析
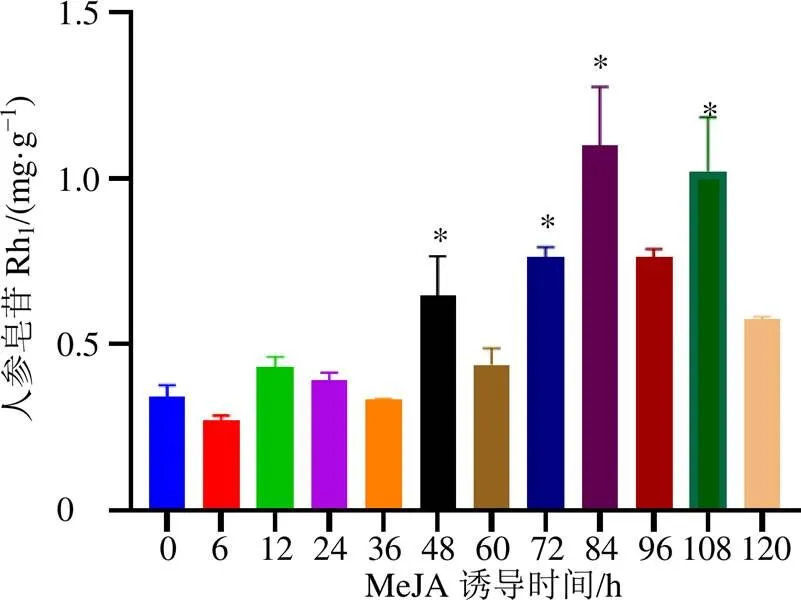
与对照组(0 h)比较:*P<0.05
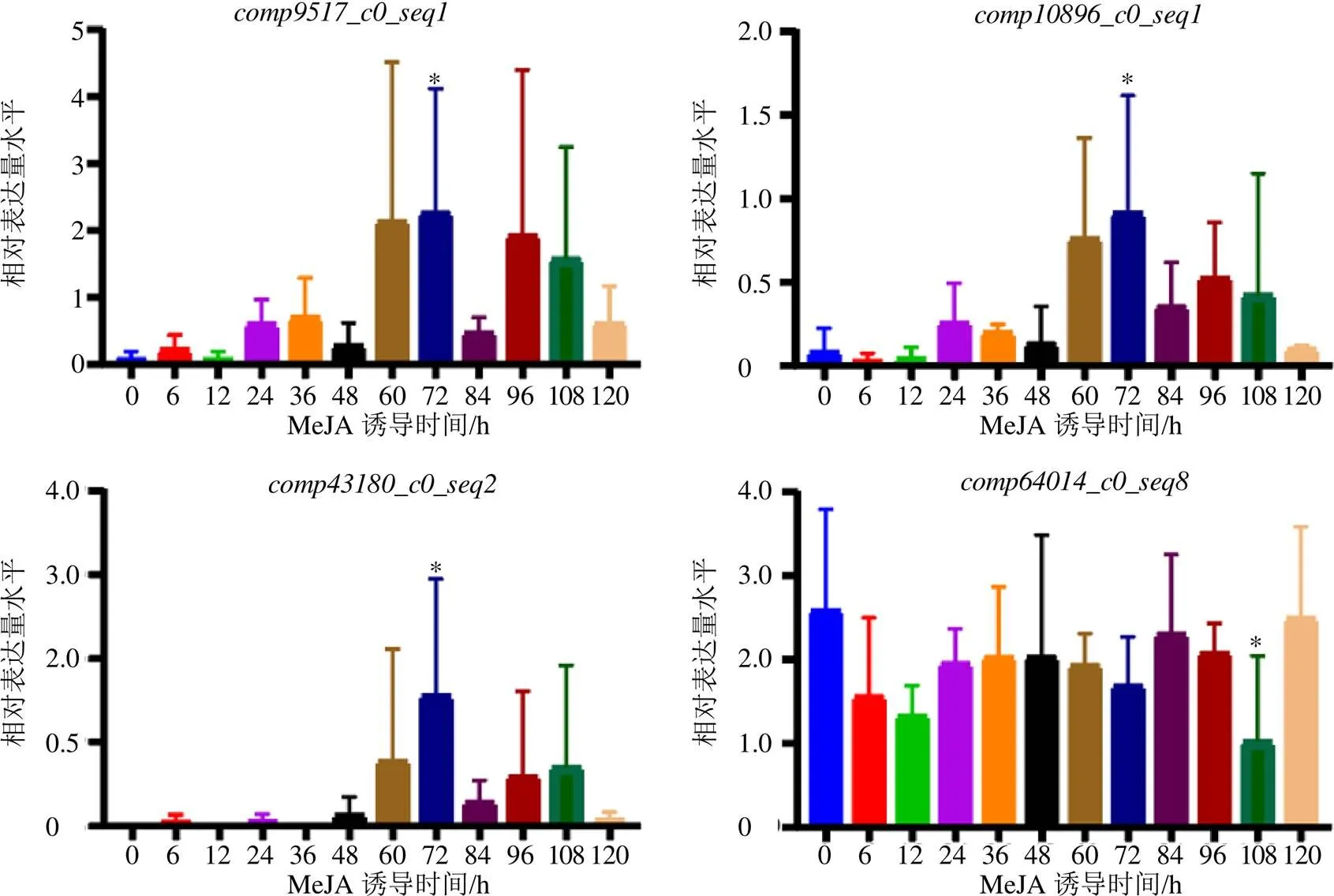
图13 人参皂苷Rh1合成候选基因在MeJA处理下的表达分析
4 讨论
由于生物系统的复杂性,许多关键基因在植物次级代谢物的生成过程中尚未明确,这些基因广泛分布在植物染色体中,因此挖掘与表征它们具有一定的挑战性。人参中的主要活性成分,人参皂苷的合成路径及其机制,一直是人参研究的关键话题。
目前已经对基因家族[27]、基因家族[28]、基因家族[29]、基因家族[30]、P基因家族[31]以及基因家族[32]等在人参皂苷生物合成过程中起作用的多个基因家族进行了全面的分析和研究,这极大地加强了对人参皂苷合成机制的理解,并为其提供了宝贵的基因资源。
随着高通量测序技术的不断突破与创新和成本的不断降低,多样本的转录组测序在系统研究生命科学问题中已被广泛应用。然而,如何从这些庞大数据中挖掘具有生物学意义的信息已成为转录组分析的核心问题[33-34]。在这种背景下,涌现出了大量的生物信息分析工具和方法。WGCNA作为一种系统生物学方法,可以阐述基因在不同样本间关联模式。WGCNA将大量的基因分组为若干模块,这些模块内的基因表达模式具有相同的表达模式。通过与目标性状进行相关性分析确定关键模块,并根据连通性筛选模块内的核心基因,以此可以快速定位与目标表型显著关联的基因。对共表达基因模块的鉴定、与表型性状的关联以及关键枢纽基因的定位具有重要意义[35]。
Yao等[36]对重组自交系的WDD01514(E1)、ZYD00463(E2)及其重组自交系的后代(E23和E171)在大豆发育的2个关键时期进行RNA-seq、WGCNA等分析鉴定出6个与种子含油量和种子重量相关的模块后,并结合后续生物信息学分析、q-PCR等验证6个可能参与种子形成和含油量积累的关键基因,为提高大豆的产量和种子油量提供理论支持。Zhu等[37]利用RNA-seq和差异基因分析,在水稻转录组的28 432个表达基因中鉴定出457个响应盐胁迫的核心基因,进一步利用WGCNA分析、GO功能注释、KEGG分析以及qRT-PCR鉴定得到编码、、、、、和转录因子等不同家族蛋白的重要枢纽基因,推动了培养水稻耐盐新品种的研究。Li等[38]对C2P5A和C2P5B 2种不同的棉花品种在发育的3个不同阶段(花粉母细胞、四分体和单核阶段)进行RNA-seq、WGCNA分析、差异基因分析等,挖掘得到3个与细胞质雄性不育(CMS)高度相关的模块和7个与生长发育相关的基因,为棉花育种和生产提供依据。
次生代谢物的生物合成与调控构建了一个多层次的网络,这需要多种关键诱导子(如MeJA和水杨酸)和信号通道的合作。已有证据确认,MeJA是一种强大的诱导子,能够在多种药用植物中刺激高价值药用次生代谢物的生产[39]。对于人参培养细胞和人参不定根,MeJA发挥了显著作用,增加了人参皂苷含量,并且能够上调相关的代谢酶基因表达[40-41]。Kim等[42]发现,MeJA能诱发、等基因的表达,同时也刺激了皂苷的积累。因此,在本研究中选择使用MeJA诱导处理人参不定根,并进行了深入分析。
目前,人参皂苷生物合成关键酶基因,如[43-45]、[46]、[47]、[48-50]、[51],均响应MeJA调控,皂苷含量和基因表达量都发生了变化,以此证明这些基因参与人参皂苷的生物合成。
本研究结果显示,可能的人参皂苷Rh1皂苷候选基因、、、在MeJA诱导下表达量均发生显著变化,但只有的表达趋势与人参皂苷Rh1在MeJA诱导下的表达趋势相同,表明这条候选基因可能与人参皂苷Rh1的合成密切相关。
总的来说,通过WGCNA分析方法,挖掘出了与人参皂苷Rh1含量高度相关的关键基因模块,成功地鉴定了与人参皂苷Rh1生物合成途径相关的关键候选基因。这些结果为进一步研究人参皂苷Rh1的生物合成机制提供了重要的线索,也为人参的遗传改良和代谢工程提供了可能的目标。
通过对304个人参样本中人参皂苷Rh1含量进行分组,利用差异基因分析的方法成功获得703个差异基因。基于304份材料中703个差异基因的表达量与人参皂苷Rh1的含量,通过WGCNA分析共获得6个基因表达模块。通过模块与表型的关联分析结果显示Green模块与人参皂苷Rh1含量的关联性最高。对Green模块进行GO富集分析和KEGG富集分析结果显示Green模块中的基因可以富集到细胞代谢、RNA结合和细胞质修饰等GO term;同时参与碳水化合物代谢、转运、信号转导、运输和分解代谢等途径。因此,Green模块中的基因参与植物代谢产物的合成。通过Cytoscape软件对Green模块内的基因进行了可视化处理,并筛选出了7个核心基因。通过查询韩国人参基因组注释。结果显示7条核心基因注释到了人参皂苷合成基因和生长素响应因子等生物功能。
采用MeJA诱导处理人参不定根后,发现、、、4条合成候选基因的表达量变化趋势只有与人参皂苷Rh1在MeJA诱导处理下的表达量变化趋势相同,因此这条基因可能在人参皂苷Rh1皂苷合成过程中发挥重要作用。
利益冲突 所有作者均声明不存在利益冲突
[1] Ratan Z A, Haidere M F, Hong Y H,. Pharmacological potential of ginseng and its major component ginsenosides [J]., 2021, 45(2): 199-210.
[2] 孙小单, 王天鸣, 李慧, 等. 人参皂苷Rh2抑制人非小细胞肺癌细胞增殖的机制研究[J]. 中草药, 2022, 53(2): 441-448.
[3] 毕云枫, 姜珊, 郑明珠, 等. 稀有原人参三醇型皂苷的生物转化研究进展 [J]. 中草药, 2017, 48(19): 4120-4125.
[4] Jin Y J, Tangchang W, Kwon O S,. Ginsenoside Rh1ameliorates the asthma and allergic inflammation via inhibiting Akt, MAPK, and NF-κB signaling pathwaysand[J]., 2023, 321: 121607.
[5] Yoon J H, Choi Y J, Lee S G. Ginsenoside Rh1suppresses matrix metalloproteinase-1 expression through inhibition of activator protein-1 and mitogen-activated protein kinase signaling pathway in human hepatocellular carcinoma cells [J]., 2012, 679(1/2/3): 24-33.
[6] Su W Y, Fan M L, Li Y,. 20()-ginsenoside Rh1alleviates T2DM induced liver injury via the Akt/FOXO1 pathway [J]., 2022, 20(9): 669-678.
[7] Yang J L, Hu Z F, Zhang T T,. Progress on the studies of the key enzymes of ginsenoside biosynthesis [J]., 2018, 23(3): 589.
[8] Hou M Q, Wang R F, Zhao S J,. Ginsenosides ingenus and their biosynthesis [J]., 2021, 11(7): 1813-1834.
[9] Yin J X, Zhang D H, Zhuang J J,. Study on the correlation between gene expression and enzyme activity of seven key enzymes and ginsenoside content in ginseng in over time in Ji’an, China [J]., 2017, 18(12): 2682.
[10] Velculescu V E, Zhang L, Zhou W,. Characterization of the yeast transcriptome [J]., 1997, 88(2): 243-251.
[11] 李欣, 李小俊, 陈晓丽, 等. 转录组数据分析与功能基因挖掘 [J]. 畜牧兽医学报, 2019, 50(3): 474-484.
[12] 章鲁瑶, 张洋, 张书筠, 等. 类风湿关节炎免疫相关差异表达基因分析 [J]. 牡丹江医学院学报, 2023, 44(1): 44-50.
[13] Chaudhary S, Sharma P C. DeepSAGE based differential gene expression analysis under cold and freeze stress in seabuckthorn (L.) [J]., 2015, 10(3): e0121982.
[14] 吴萍, 王晓宇, 李青苗, 等. 基于高通量转录组测序的白芷差异表达基因分析 [J]. 西南农业学报, 2020, 33(2): 233-240.
[15] 肖丽君, 陈谦明, 吴玉琪, 等. 基于转录组测序分析芍药苷诱导下口腔黏膜上皮细胞差异表达基因 [J]. 口腔医学研究, 2023, 39(6): 527-533.
[16] Chang Y J, Wang M Z, Li J,. Transcriptomic analysis reveals potential genes involved in tanshinone biosynthesis in[J]., 2019, 9(1): 14929.
[17] Lei H Y, Niu T Z, Song H F,. Comparative transcriptome profiling reveals differentially expressed genes involved in flavonoid biosynthesis between biennial and triennial[J]., 2021, 161: 113217.
[18] Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis [J]., 2008, 9: 559.
[19] Liu W, Li L, Ye H,. Weighted gene co-expression network analysis in biomedicine research [J]., 2017, 33(11): 1791-1801.
[20] Liu D F, Wang Z J, Zhong L,. Targets and potential mechanism ofin treatment of primary hepatocellular carcinoma based on bioinformatics analysis [J]., 2022, 2022: 8762717.
[21] Zeng G P, Li Z, Zhao Z. Analysis of weighted gene co-expression network of triterpenoid-related transcriptome characteristics from different strains of[J]., 2021, 11(1): 18207.
[22] 邬东东, 杨启帆, 方柔柔, 等. 基于网络药理学联合加权基因共表达网络分析黄精治疗代谢性相关脂肪性肝病的作用机制 [J]. 现代药物与临床, 2023, 38(7): 1606-1614.
[23] Liu S Z, Jiang Y, Wang Y F,. Genetic and molecular dissection of ginseng (Mey.) germplasm using high-density genic SNP markers, secondary metabolites, and gene expressions [J]., 2023, 14: 1165349.
[24] Kim N H, Jayakodi M, Lee S C,. Genome and evolution of the shade-requiring medicinal herb[J]., 2018, 16(11): 1904-1917.
[25] 于靖辉. 人参内生菌广泛靶向代谢组学分析及其对人参不定根的影响 [D]. 长春: 吉林农业大学, 2023.
[26] Jiang Y, Liu S Z, Li L,. Transcriptome and phenotype integrated analysis identifies genes controlling ginsenoside Rb1biosynthesis and reveals their interactions in the process in[J]., 2022, 23(22): 14016.
[27] Liu Q, Sun C Y, Han J Z,. Identification, characterization and functional differentiation of thegene family and its roles in response to cold stress in ginseng,C.A. Meyer [J]., 2020, 15(6): e0234423.
[28] Chen J, Zhou Y H, Zhang Q,. Structural variation, functional differentiation and expression characteristics of the AP2/gene family and its response to cold stress and methyl jasmonate inC.A. Meyer [J]., 2020, 15(3): e0226055.
[29] Zhu L, Zhao M Z, Chen M Y,. Thegene family and its response to saline stress in Jilin ginseng,C.A. Meyer [J]., 2020, 295(4): 877-890.
[30] Li H J, Chen J, Zhao Q,. Basic leucine zipper () transcription factor genes and their responses to drought stress in ginseng,C.A. Meyer [J]., 2021, 22(1): 316.
[31] Wang Y, Li X Y, Lin Y P,. Structural variation, functional differentiation, and activity correlation of the cytochrome P450 gene superfamily revealed in ginseng [J]., 11: 170106.
[32] Wang N, Wang K Y, Li S K,. Transcriptome-wide identification, evolutionary analysis, and GA stress response of thegene family inC. A. Meyer [J]., 2020, 9(2): 190.
[33] Tian J D, Ma K S, Saaem I. Advancing high-throughput gene synthesis technology [J]., 2009, 5(7): 714-722.
[34] 王艳花, 刘景森, 李加纳. 整合GWAS和WGCNA筛选鉴定甘蓝型油菜生物产量候选基因 [J]. 作物学报, 2021, 47(8): 1491-1510.
[35] 龙佳丽, 邹奕, 邳植, 等. 转录组测序揭示激素介导的信号通路参与甜菜低温应答 [J]. 中国农学通报, 2021, 37(31): 5-14.
[36] Yao Y J, Xiong E H, Qu X L,. WGCNA and transcriptome profiling reveal hub genes for key development stage seed size/oil content between wild and cultivated soybean [J]., 2023, 24(1): 494.
[37] Zhu M D, Xie H J, Wei X J,. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice [J]., 2019, 10(9): 719.
[38] Li Y Q, Qin T F, Wei C Y,. Using transcriptome analysis to screen for key genes and pathways related to cytoplasmic male sterility in cotton (L.) [J]., 2019, 20(20): 5120.
[39] Ho T T, Murthy H N, Park S Y. Methyl jasmonate induced oxidative stress and accumulation of secondary metabolites in plant cell and organ cultures [J]., 2020, 21(3): 716.
[40] Lu J, Li J X, Wang S H,. Advances in ginsenoside biosynthesis and metabolic regulation [J]., 2018, 65(4): 514-522.
[41] Cao H Z, Nuruzzaman M, Xiu H,. Transcriptome analysis of methyl jasmonate-elicitedadventitious roots to discover putative ginsenoside biosynthesis and transport genes [J]., 2015, 16(2): 3035-3057.
[42] Kim O T, Bang K H, Kim Y C,. Upregulation of ginsenoside and gene expression related to triterpene biosynthesis in ginseng hairy root cultures elicited by methyl jasmonate [J]., 2009, 98(1): 25-33.
[43] Kim Y J, Zhang D B, Yang D C. Biosynthesis and biotechnological production of ginsenosides [J]., 2015, 33(6 Pt 1): 717-735.
[44] Lee M H, Jeong J H, Seo J W,. Enhanced triterpene and phytosterol biosynthesis inoverexpressing squalene synthase gene [J]., 2004, 45(8): 976-984.
[45] Kim T D, Han J Y, Huh G H,. Expression and functional characterization of three squalene synthase genes associated with saponin biosynthesis in[J]., 2011, 52(1): 125-137.
[46] Kim Y K, Kim Y B, Uddin M R,. Enhanced triterpene accumulation inhairy roots overexpressing mevalonate-5-pyrophosphate decarboxylase and farnesyl pyrophosphate synthase [J]., 2014, 3(10): 773-779.
[47] Han J Y, Kwon Y S, Yang D C,. Expression and RNA interference-induced silencing of the dammarenediol synthase gene in[J]., 2006, 47(12): 1653-1662.
[48] Chen S, Luo H, Li Y,. 454 EST analysis detects genes putatively involved in ginsenoside biosynthesis in[J]., 2011, 30(9): 1593-1601.
[49] Yan X, Fan Y, Wei W,. Production of bioactive ginsenoside compound K in metabolically engineered yeast [J]., 2014, 24(6): 770-773.
[50] Wang P P, Wei Y J, Fan Y,. Production of bioactive ginsenosides Rh2and Rg3by metabolically engineered yeasts [J]., 2015, 29: 97-105.
[51] Han J Y, Hwang H S, Choi S W,. Cytochrome P450 CYP716A53v2 catalyzes the formation of protopanaxatriol from protopanaxadiol during ginsenoside biosynthesis in[J]., 2012, 53(9): 1535-1545.
Mining of genes related to biosynthesis of ginsenoside Rh1 based on WGCNA in
LIU Chang, LIU Sizhang, YU Jinghui, CHEN Ping, LEI Jun, WANG Yi, ZHANG Meiping
Jilin Province Engineering Research Center of Ginseng Gene Resources Development and Utilization,College of Life Science, Jilin Agriculturial University, Changchun, 130118, China
The aim of this study is to unearth genes associated with the biosynthesis of ginsenoside Rh1employ through weighted gene co-expression network analysis (WGCNA).Utilizing transcriptome sequencing data from 304 distinct domesticated varieties of ginseng cultivated in the primary ginseng-producing region of Jilin Province, China, we conducted a differentially expressed gene mining with ginsenoside Rh1content as the phenotypic parameter.A total ofsix co-expression modules of genes intimately correlated with ginsenoside Rh1content was gained by employing WGCNA. Subsequent correlation analyses highlighted the Green module as a pivotal module significantly associated with ginsenoside Rh1content. Functional annotation results revealed that genes within the Green module were enriched in several pathways, including mRNA binding and protein modification. A total of seven core genes were selected by constructing gene interaction network, and functional prediction indicated that these genes may play crucial roles in the biosynthesis pathway of ginsenoside Rh1.modulation analysis of methyl jasmonate (MeJA) was performed on these seven candidate genes. Upon MeJA induction in ginseng adventitious roots, the expression levels of,,, andand ginsenoside Rh1content exhibited significant simultaneous changes. However, only the expression trend of compwas the same as that of ginsenoside Rh1induced by MeJA, indicating that this candidate gene may be closely related to ginsenoside Rh1synthesis.This study lays the theoretical foundation for unraveling the biosynthesis pathway of ginsenoside Rh1.
C. A. Mey.; ginsenosides Rh1; differentially expressed gene; WGCNA; methyl jasmonate
R286.12
A
0253 - 2670(2024)08 - 2723 - 11
10.7501/j.issn.0253-2670.2024.08.021
2023-10-05
吉林省科技厅项目(20180101027JC,20200403011SF);国家自然科学基金青年基金项目(31401445)
刘 畅,硕士研究生,研究方向为人参基因组学。Tel: 15148182822 E-mail: 2454278361@qq.com
通信作者:张美萍,博士生导师,研究方向为人参功能基因组学、人参分子设计育种。Tel: 13504315977 E-mail: meiping.zhang@jlau.edu.cn
[责任编辑 时圣明]
