CaMKIIγ和CaMKIIδ通过PI3K/Akt/Erk 信号通路减轻小鼠神经元缺血再灌注损伤
2024-04-13刘昊铭林子诗
刘昊铭,林子诗,叶 靖
1南方医科大学南方医院麻醉科,广东 广州 510515;2佛山市第一人民医院麻醉科,广东 佛山 528000;3南方医科大学珠江医院麻醉科,广东 广州510260
缺血性脑卒中是全球范围内致残率最高的疾病之一,也是导致患者死亡的第3大原因[1]。由于人口基数大以及老龄化,我国缺血性脑卒中患者数居全球首位[2]。尽快恢复脑卒中患者缺血区域血流灌注有助于神经细胞功能修复,但会诱发脑缺血再灌注损伤,造成继发性神经功能恶化。目前针对脑缺血再灌注损伤的确切治疗手段尚有待开发[3]。
钙/钙调节蛋白依赖蛋白激酶II(CaMKII)家族是一种丝氨酸/苏氨酸特性的多功能蛋白激酶,通过钙离子信号通路调节多种细胞的生存与活性,参与调节突触可塑性,影响行为、海马学习及记忆功能;在脑缺血中,CaMKII表达和/或激活的改变是缺血诱导神经细胞死亡的关键因素[4-6]。但既往关于CaMKII 对缺血性脑卒中神经保护的研究结果颇具争议。有报道指出CaMKII具有促进凋亡的神经损伤作用:CaMKII过表达可增加谷氨酸诱导的缺血损伤后神经元死亡率,使用CaMKII抑制剂则可减轻缺血后神经损伤[7-9]。另一些研究表明,CaMKII可以通过磷酸化和抑制促凋亡蛋白一氧化氮合酶、Bax等发挥神经保护作用;激活CaMKII可提高缺血缺氧神经元的存活率,敲除CaMKII基因可导致更严重的神经细胞死亡[4,10-12]。造成上述对立观点的原因除了研究使用了非特异性CaMKII抑制剂、神经元来源于不同脑区或不同生长时期、缺血后损伤的观察时点不一之外[4,13],更重要的是既往研究并未区分CaMKII各同工型对缺血靶点的影响。
CaMKⅡ包括α、β、γ、δ 4类同工型,课题组前期研究证实缺血再灌注神经元细胞对CaMKII各同工型的表达差异,在针对单个同工型的探讨中发现:CaMKIIδ和CaMKIIγ通过激活核转录因子κB(NF-κB)信号通路产生神经保护作用[14,15]。然而在不同的缺血再灌注神经元研究中,NF-κB信号通路会表现出促进炎症凋亡作用或者保护作用[10,14-16]。因此我们猜测存在其他途径参与该通路介导CaMKIIδ和CaMKIIγ的神经保护作用。
细胞外信号调节激酶(Erk)信号途径和磷脂酰肌醇-3-激酶/蛋白激酶B(PI3K/Akt)信号途径两个通路均可因细胞缺血缺氧而被激活,影响并调控下游的NF-κB信号通路[17]。但目前尚无研究探讨CaMKIIδ和CaMKIIγ的神经保护作用与PI3K/Akt/Erk 通路的内在联系。本研究拟在前期研究基础上,通过氧糖剥夺/复氧(OGD/R)和大脑中动脉闭塞(MCAO)模拟缺血再灌注,探讨PI3K/Akt/Erk 是否参与调控CaMKIIγ和δ对脑缺血再灌注损伤的保护机制。
1 材料和方法
1.1 材料
1.1.1 实验试剂GAPDH 抗体(Badrila Inc);CaMKII(pan)(D11A10)抗体(Cell Signaling);p44/42MAPK(Ek1/2)抗体、Phospho-p4/42MAPK(Erkl/2)抗体、Akt抗体、Phospho-Akt(Ser473)抗体(Cell Signaling);胎牛血清(Gib-ol);0.05%和0.25%胰酶、2,3,5氯化三苯基四氮唑(Sigma);碘化丙啶、DharmaFECT 转染试剂(Thermofish);钙黄绿素(Invitrogen);CAMK2G 和CAMK2D-小干扰RNA(si-RNA)和非靶向siRNA(si-NT)(Integrated DNA Technologies);P3 Primary Cell 4d-nucleofector kits(Lonza)。
1.1.2 动物 SPF级雄性的BALB/e小鼠18~22 g 8~10周、孕18 d的野生型SD大鼠50只,购自南方医科大学动物实验中心[质量合格许可证号SCXK(粤)2016-0041],洁净级饲养环境,在恒温26 ℃条件下,以12 h光照-黑暗交替循环。所有实验步骤均按南方医科大学实验动物伦理委员会批准开展(伦理批号:LAEC-2021-074)。
1.2 方法
1.2.1 胎鼠皮质神经元细胞分离培养
1.2.1.1 神经元提取 把SD孕鼠放入吸入麻醉诱导装置,以3%七氟醚与空气混合充入诱导箱中。待其进入麻醉状态后,迅速将胚胎剖出,并浸泡在预先上预冷的75%酒精内消毒。使用灭菌消毒后的剪刀迅速将胎鼠断头,分离胎鼠大脑并迅速将其置于事先预冷好的装有DMEM-F12 的培养皿中。剪碎大脑组织后加入0.125%的胰酶,等待15 min后加入含有10%胎牛血清的DMEM-F12 完全培养基。使用移液枪轻柔吹打3次,后静置1 min后弃掉上清。加入含有10 μm/mLDNA酶的完全培养基,反复吹打使神经元分离到培养基中,静置1 min后吸取上层清液,滴入盖有孔径40 μm滤膜的40 mL离心管中。加入DMEM-F12,重复以上操作2次。将收集到的所有细胞于4 ℃、1000 r/min离心5 min,吸出上层清液,用神经元接种液(DMEM-F12基础培养基+10%胎牛血清+1%的青霉素链霉素溶液)重悬细胞[18]。
1.2.1.2 接种神经元 预先按每孔1 mL的量向6孔板中加入多聚赖氨酸(0.1 mg/mL)。消毒后放置于37 ℃培养箱中过夜。次日稀释神经元悬液到需要浓度(1×105、5×105、1×106细胞/mL)分别加入1 mL六孔板的上样孔中,培养环境为37 ℃、5%CO2、饱和湿度,等待原代神经元充分贴壁。
1.2.2 si-RNA 转染 根据产品说明使用P3 Primary Cell 4D-Nucleofector试剂对原代神经元细胞进行Myc-DDK-CaMKIIδ质粒(OriGene Technologies)转染。使用Dharma FECT转染试剂和Cell Line Nucleofector V试剂将CAMK2GsiRNA(si-CAMK2G)、CAMK2DsiRNA(si-CAMK2D)和非靶向siRNA(si-NT)转染到原代神经元细胞中。转染48 h后,原代神经元接受OGD/R处理,然后在常氧培养条件下复氧不同时间,并观察Erk、磷酸化Erk(p-Erk)、Akt、磷酸化Akt(pS473-Akt)的表达。
1.2.3 OGD/R模型 在对37℃、5%CO2条件中培养的神经元细胞更换无糖培养液后,将其置入1% O2+5%CO2+94%N2的37 ℃缺氧箱内培养1 h;之后以原培养方式继续观察72 h。对照组细胞则一直放置于37 ℃、5%CO2培养箱内,不更换无糖培养液。通过钙黄绿素AM/碘化丙啶染色评估细胞生存率[19]。
1.2.4 小鼠MCAO模型建立[19]随机将小鼠分为假手术组(Sham)和MCAO组,每组各25只,使用3%七氟醚混合空气诱导小鼠进入麻醉状态后用1%七氟醚维持,在显微镜下分离颈部组织暴露左侧颈总动脉、颈外动脉及颈内动脉,Sham组只进行动脉分离。MCAO组使用4-0号缝合线行颈总动脉的近心端和颈外动脉的远心端结扎;由颈总动脉将头端直径0.23 mm、主干直径0.18 mm 的尼龙栓线插入至大脑中动脉,深度约120 mm,阻断1 h后抽出线栓,缝合并消毒术口后将小鼠放入笼中饲养,术后24 h后对两组小鼠进行神经行为学评分,以此判断MCAO 模型的效果,神经功能缺陷评分标准如下[19]:0=无可观察的缺陷;1=前肢屈曲;2=前肢屈曲和侧推抗力减弱;3=前肢屈曲、侧推抗力减弱和单侧盘旋;4=前肢屈曲和行走能力受损或丧失。取大脑组织行2,3,5-三苯基氯化四氮唑染色,应用Image J软件评估脑梗死面积。并于再灌注后24、48、72和96 h观察梗死灶周围脑组织的CaMKIIγ和CaMKIIδ、Erk、p-Erk、Akt和pS473-Akt的表达。
1.2.5 Western blot检测 将200 μL RIPA细胞裂解液分别加入神经元样品中,30 min后将裂解好的神经元碎片混合液离心5 min(4 ℃1000 r/min)。收集上清液使用BCA蛋白测定试剂盒定量蛋白裂解物。各取30 μg 样品经过SDS-聚丙烯酰胺凝胶电泳分离后转移至PVDF膜上。PVDF 膜取出放置在配好的封闭液中(10 mL TBST+脱脂奶粉0.2 g),在室温下封闭2 h。一抗(1∶1000稀释)4 ℃孵育过夜,二抗(1∶1000稀释)常温孵育1 h。结果通过化学发光进行可视化。使用Imaging J软件对图像进行定量分析。
1.3 统计学分析
计量资料采用均数±标准差表示,多组间的比较采用ANOVA 检验。使用SPSS26.0 统计软件进行统计分析,P<0.05被认为差异有统计学意义。
2 结果
2.1 敲减CaMKⅡγ显著降低大鼠神经元细胞的生存率
与转染si-NT的对照细胞相比,转染si-CAMK2G的神经元细胞在OGD/R 12、24、48、72 h后,多数细胞形态呈现出梭形或圆形,细胞体积缩小,突触数目减少(图1)。细胞生存率明显低于si-NT(图2)。

图1 OGD/R后0~72 h钙黄绿素AM/碘化丙啶染色的原代神经元图像Fig.1 Representative images of calcein-AM/propidium iodide(PI)staining of the primary neurons from 0 h to 72 h following oxygen-glucos deprivation/reperfusion (OGD/R) (Original magnification: ×200).The red channel shows PI staining for cell death,and the green shows calcein-AM staining of live cells.

图2 敲减CAMK2G可增加OGD/R诱导的原代神经元细胞死亡Fig.2 Knockdown of CAMK2G exacerbates OGD/Rinduced neuronal cell death.Cell survival was assessed by calcein-AM/PI staining.The total PI-positive or calcein-AMpositive cells were counted from 10 random fields in each image.The neuron survival rate was calculated as the ratio of calcein-AM-positive cell number over the total cell number by Image J.**P<0.01,***P<0.001.
2.2 敲减CAMK2G或CAMK2D可抑制PI3K/Akt/Erk信号通路表达上调
与对照组细胞相比,转染si-CAMK2G可特异性逆转OGD/R 介导的鼠神经元细胞CaMKIIγ 的表达上调,转染si-CAMK2D可特异性逆转CaMKIIδ的表达上调(P<0.01)。与对照组细胞相比,转染si-CAMK2G或si-CAMK2D可使Erk、p-Erk和Akt的蛋白表达明显受到抑制;转染si-CAMK2D1 可抑制pS473-Akt的蛋白表达(P<0.01)。与转染了si-NT的胎鼠原代神经元细胞相比,转染si-CAMK2G或si-CAMK2D可抑制pS473-Akt的蛋白表达(P<0.01,图3、4)。

图3 OGD/R对si-NT、si-CAMK2D、si-CAMK2G转染后的大鼠原代神经元细胞中PI3K/Ark/Erk通路组件的影响Fig.3 Knockdown of CAMK2D or CAMK2G inhibits upregulation of CaMKIIδ or CaMKIIγ,respectively,and suppresses the expressions of Erk,p-Erk,Akt and pS473-Akt induced by OGD/R.GAPDH was blotted as the loading control.Representative images from 3 independent experiments are shown.

图4 各灰度值表达量化图Fig.4 Quantification of the expression levels of CaMKIIδ,CaMKIIγ,Akt,phosphorylate-Akt,Erk and phosphorylate-Erk in the neurons with or without CAMK2G and CAMK2D knockdown by densitometry analysis.Percent control(y-axis)represents the expression of the target genes to that of the controls under normoxic condition(100%).Con:Control.p-Erk:Phosphorylate-Erk.Data are Mean±SD from 4 independent experiments.**P<0.01 vs control.
2.3 MCAO増加小鼠脑CaMKIIδ和CaMKⅡγ的表达,激活PI3K/Akt/Erk信号通路
MCAO组小鼠缺血后出现100%运动减少、90%侧倾姿势、84%扁平姿势、转圈68%、低反应性52%、前肢屈曲100%、肌力下降100%、运动失调100%神经行为学总体评分增加,而sham组无明显变化MCAO诱导的脑组织梗死会在24 h 内显现,缺血脑区的神经元进行性坏死会在72 h达到峰值,随后在96 h出现一定恢复(图5)。从梗死灶周围的半暗区域收集的脑组织,使用CaMKII(pan,D11A10)抗体通过Western blot 可检测到CaMKIIδ和CaMKIIγ在24、48和72 h持续上调,并在96 h 回调,CaMKIIδ显著高于sham 组再灌注24 h(P<0.0001),CaMKIIγ(P<0.05或0.01);sham组内各观察时点的CaMKIIδ和CaMKIIγ表达与再灌注24 h比较差异无统计学意义(P>0.05,图6、7)。
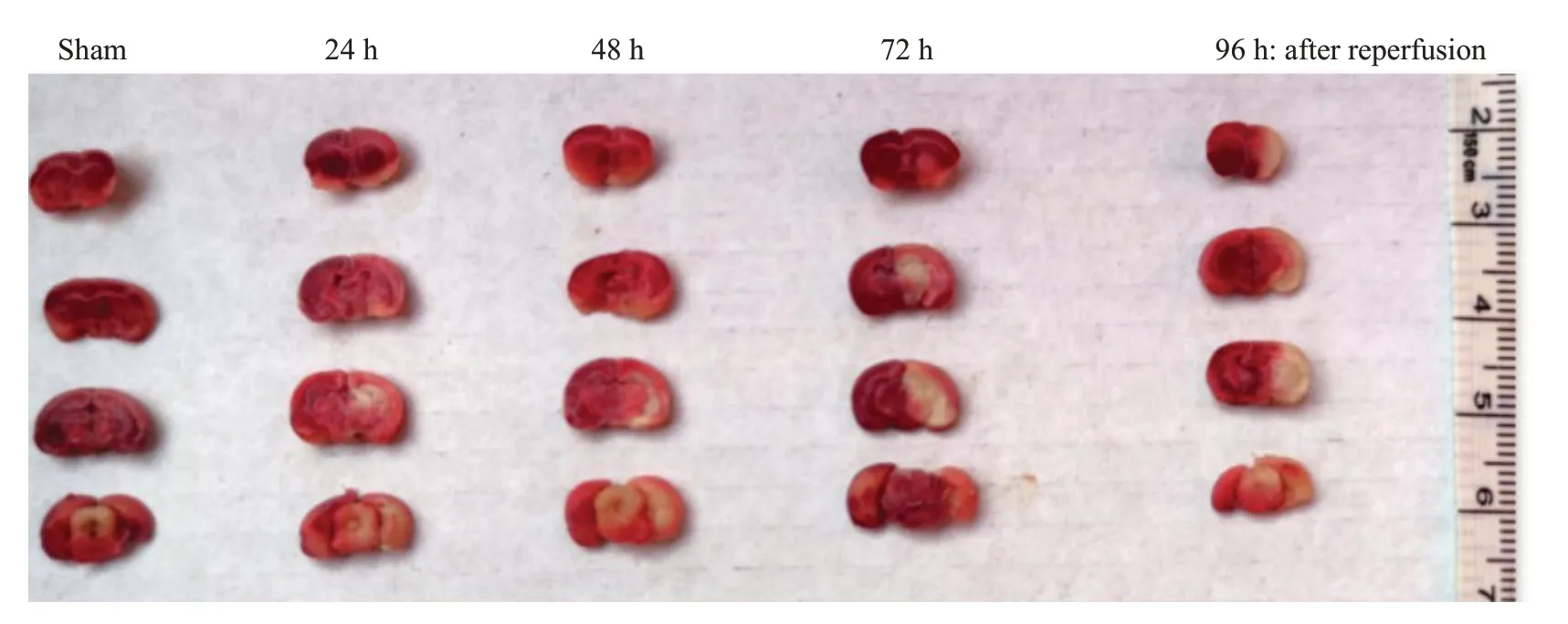
图5 MCAO 诱导小鼠脑梗死Fig.5 Cerebral infarction in mice induced by MCAO observed at 24,48,72,and 96 h after reperfusion.
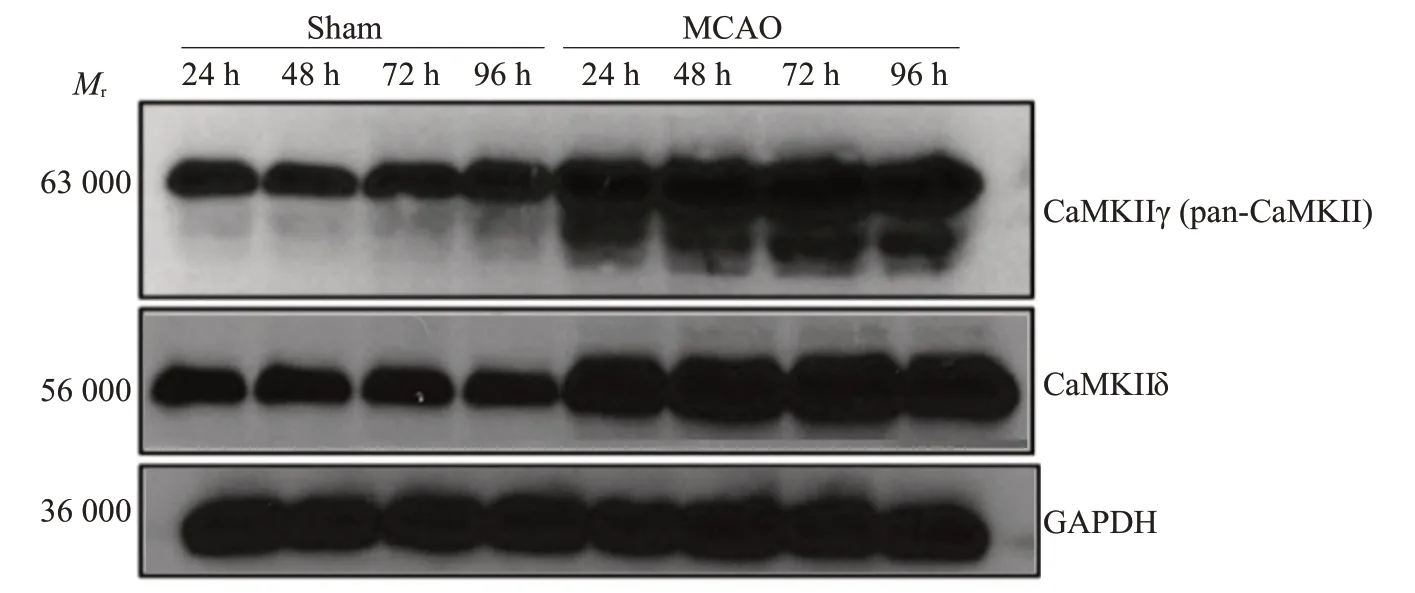
图6 通过Western blotting分析两组梗死灶周围脑组织的CaMKIIγ和CaMKIIδ的表达Fig.6 Analysis of CaMKIIγ and CaMKIIδ expressions in the penumbra of sham-operated and MCAO mice at 24,48,72 and 96 h after reperfusion by Western blotting.
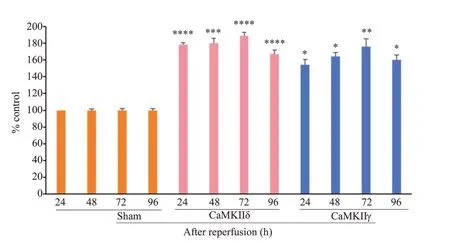
图7 通过灰度值分析将CaMKIIδ和CaMKIIγ 的表达量化Fig.7 Quantification of the levels of CaMKIIδ and CaMKIIγ by densitometry analysis.The value in sham-operated group at 24 h served as the control(100%).Data are Mean±SD from 4 independent experiments.*P<0.05;**P<0.01;***P<0.001;****P<0.0001 vs sham 24 h.
在MCAO 造模成功再灌注24、48、72 和96 h 后,Erk、p-Erk、Akt、pS473-Akt的表达均高于sham组再灌注24 h 的表达(P<0.05 或0.01),上调变化趋势与CaMKIIδ和CaMKIIγ相似;sham组内各观察时点Erk、p-Erk、Akt、pS473-Akt的表达与再灌注24 h比较差异无统计学意义(P>0.05,图8、9)。

图8 Sham组和MCAO组织梗死灶周围组织中PI3K/Akt/Erk 信号通路的Western blot分析Fig.8 PI3K/Akt/Erk signaling pathway in the penumbra of sham-operated and MCAO mice at 24,48,72 and 96 h after reperfusion by Western blotting.Representative images from 4 independent experiments are shown.

图9 各灰度值表达量化表Fig.9 Quantification of the levels of Akt,phosphorylate-Akt,Erk and phosphorylate-Erk in the penumbra of sham-operated and MCAO mice by densitometry analysis.Data are Mean±SD from 4 independent experiments.*P<0.05;**P<0.01 vs sham 24 h.
3 讨论
课题组前期研究使用能识别所有CaMKII同工型的pan CaMKII抗体,首次证明在原代神经元中,缺血再灌注损伤可选择性地诱导CaMKIIδ和CaMKIIγ[14,15]。课题组发现在缺血再灌注后,小鼠来源神经瘤母细胞中主要上调的同工型CaMKIIδ,而非CaMKIIα或CaMKIIβ。相比之下,在小鼠原代皮质神经元中,CaMKIIγ是上调程度最高的同工型,其次是CaMKIIδ[15]。CaMKIIα和CaMKIIβ在离体缺氧损伤神经元中的表达远低于CaMKIIδ 和CaMKIIγ,且缺血 再灌注 损伤诱导的CaMKIIγ高表达及持续上调是一个新的发现,暗示了这种同工型在神经元中的潜在重要作用。此外,CAMK2G 在DNA 水平的峰值上调达7 倍,远高于CAMK2D的3~4倍,表明CaMKIIγ可能对大脑中的神经元具有重要的功能。此外,我们还证实缺血再灌注的神经元细胞对CaMKII各同工型的表达差异,并通过质粒转染过表达以及si-RNA 敲减CAMK2D 和CAMK2G证明其通过激活NF-κB信号通路产生神经保护作用,显示了NF-κB信号通路对缺血再灌注神经元不仅具有促进炎症凋亡作用,还存在保护作用。
对PI3K/Akt/Erk信号通路与心脑缺血再灌注损伤的研究提示,该通路的激活与缺血再灌注损伤密切相关,由于Akt也可激活IkB激酶IKKα,使NF-κB的抑制蛋白IκB降解,进而导致NF-κB从细胞质中释放,激活其靶基因而促进细胞的存活,调节Bcl-xL,发挥抗凋亡作用。是否因为PI3K/Akt和Erk通路参与其中,才使NF-κB信号通路产生神经保护作用呢?
通过建立MCAO模型,我们发现缺血再灌注损伤可激活PI3K/Akt/Erk 信号通路:Erk、p-Erk、Akt 和pS473-Akt的表达上调。由此我们推断PI3K/Akt/Erk信号通路参与了神经缺血再灌注损伤的调控。而通过转 染si-CAMK2D 和si-CAMK2G 敲 减CAMK2D 和CAMK2G,发现PI3K/Akt/Erk信号通路组件的表达明显受到抑制。因此,我们判断CaMKIIδ和CaMKIIγ通过激活NF-κB信号通路产生神经保护作用的上游通路为PI3K/Akt/Erk 信号通 路,提示了CaMKIIδ 和CaMKIIγ在缺血性损伤期间促进神经元存活的新机制,有助于开启脑缺血再灌注损伤治疗的新思路。
此外,CaMKII四个同工型在不同的脑区表达量各不相同[20-23],而CaMKIIγ在海马和海马旁回、外侧缰核、前额叶皮质等脑区中,与学习记忆调节、认知功能、抑郁症、自闭症或精神分裂症病理生理密切相关[24-27],由此我们猜想CaMKIIγ可在脑缺血再灌注损伤神经保护的基础上,更精准地改善不同部位神经元的生存状态,促进神经功能恢复,改善相关脑区损伤后的带来的并发症。由于本系列研究均聚焦于皮质神经元,我们后续将深入探讨CaMKIIγ及其子型对不同脑区脑缺血再灌注损伤的影响,以及作用机制。
综上所述,CaMKⅡγ和CaMKIIδ在鼠皮质神经元细胞发生缺血缺氧损伤时,通过激活NF-κB信号通路产生的神经保护作用,可能由PI3K/Akt/Erk信号通路介导。
