HPLC法测定蒙药甘露解毒丸中连翘酯苷A的含量研究
2024-04-09樛跃


【摘 要】 目的:建立高效液相色谱法测定蒙药甘露解毒丸中连翘酯苷A的含量。方法:色谱柱为SHISEIDO CAPCELL PAK C18色谱柱(250 mm×4.6 mm,5 μm),以乙腈-0.4%冰醋酸(15∶85,V/V)为流动相,流速为0.8 mL/min,检测波长330nm,柱温为30 ℃,进样量为10 μL。结果:连翘酯苷A在0.1~1.4μg范围内呈现良好线性关系,线性方程为Y=2302379X+2377(r=0.9999,n=6);专属性良好;精密度和稳定性的RSD分别为0.40%和0.80%。平均加样回收率为94.34%,RSD为1.26%(n=9)。测定两批甘露解毒丸样品,平均含量为0.7772 mg/g,RSD为0.71%。结论:高效液相色谱法测定连翘酯苷A的含量操作简便,重现性好,灵敏度较高,适用于甘露解毒丸中连翘酯苷A的含量测定。
【关键词】 甘露解毒丸;连翘酯苷A;高效液相色谱法;含量测定
【中图分类号】R284.1 【文献标志码】 A【文章编号】1007-8517(2024)04-0024-04
DOI:10.3969/j.issn.1007-8517.2024.04.zgmzmjyyzz202404006
Research of Content Determination for Forsythoside A in Mongolian Medicine by HPLC
JIU Yue
Abstract:Objective To establish the high performance liquid chromatograghy method for the content determination of Forsythoside A in Mongolian medicine Ganlu Jiedu Pills. Methods The chromatographic column was SHISEIDO CAPCELL PAK C18(250 mm×4.6 mm,5 μm),the mobile phase was acetonitrile-0.4% acetic acid(15∶85,V/V), the flow rate was 0.8 mL/min, detection wavelength was 330nm, the column temperature was 30 ℃, and the injection volume was 10 μL.Results Forsythoside A showed a good linearity in the range of 0.1-1.4 μg and the linear equation was Y=2302379X+2377(r=0.9999, n=6). The specificity was good and the RSD of precision and stability were 0.40% and 0.80%. The average recovery was 94.34% with RSD of 1.26% (n=9).The average content of 2 batches samples was 0.7772 mg/g,with RSD of 0.71%. Conclusion The high performance liquid chromatograghy method for the content determination of Forsythoside A is easy to operate, reproducible and high sensitivity which can be used for the content determination of Ganlu Jiedu Pills.
Key words:Ganlu Jiedu Pills; Forsythoside A; High performance liquid chromatography; Content Determination
蒙藥制剂甘露解毒丸为棕褐色水丸,味微苦涩,由连翘、石膏、益智仁、肉豆蔻、苦参、丁香等四十七味药组成,具有清热、解毒、消食功效。用于接触性毒、阳光毒、呼吸中毒、木质性毒等各类毒性疾病[1]。方中的连翘为主药,其有效成分是以连翘苷为主的木脂素类和连翘酯苷为主的苯乙醇苷类[2]。连翘酯苷A作为一种酚性成分,一直以来都被视为连翘的主要有效成分,但其结构较为复杂,遇酸、碱或高温等不稳定,易引起分解,对其含量进行检测显得更加重要[3]。连翘酯苷A的检测方法目前有薄层色谱法、毛细管胶束电动色谱法、质谱联用法、红外光谱法、高效液相色谱法、UPLC-MS/MS法等[4-5],其中高效液相色谱法具有高速高效、高灵敏度、应用范围广等特点,已广泛应用于连翘酯苷A的含量测定[6-7]。蒙药制剂甘露解毒丸无现行质量标准,试验建立测定连翘酯苷A含量的高效液相色谱法。
1 仪器与试药
1.1 仪器 Waters e2695高效液相色谱仪(美国Waters公司);UV2700紫外-可见分光光度计[岛津仪器(苏州)有限公司];BSA124S电子天平[型号:(0.1 mg,赛多利斯科学仪器(北京)有限公司];BT125D电子天平[0.01 mg,赛多利斯科学仪器(北京)有限公司];KQ-5200DE数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药 连翘酯苷A 对照品(批号:111810-201707;中国食品药品检定研究院,纯度为97.2%);供试品:甘露解毒丸由内蒙古自治区国际蒙医医院提供(批号:20180514、20200316、20200440)。甲醇(色谱纯;批号:20201102036;山东禹王和天下新材料有限公司)、乙腈(色谱纯;批号:20200528118;山东禹王和天下新材料有限公司)、冰醋酸(色谱纯;批号:20200415;天津市大茂化学试剂厂)。
2 方法与结果
2.1 色谱条件 色谱柱:SHISEIDO CAPCELL PAK C18色谱柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.4%冰醋酸溶液(15∶85);柱温:30 ℃;检测波长:330 nm;流速:0.8 mL/min;进样量:10 μL。理论板数按连翘酯苷A计不低于3000且分离度良好,高效液相色谱图如图1所示(连翘酯苷A峰保留时间为37.21 min)。
2.2 溶液的制备
2.2.1 供试品溶液 取甘露解毒丸,研细,精密称取约1.0 g,置具塞锥形瓶中,精密加入70%甲醇25 mL,密塞,称定重量,超声(功率250 W,频率40 kHz)处理30 min,放冷,称定重量,用70%甲醇补足减失重量,摇匀,0.45 μm滤膜滤过,取续滤液,即得。
2.2.2 对照品溶液 精密称定连翘酯苷A对照品,加入甲醇制成浓度为每1 mL约含0.1 mg的溶液,0.45 μm滤膜滤过,取续滤液,即得。
2.2.3 阴性样品溶液 按“2.2.2”项下供试品溶液制备方法,制备阴性样品溶液(除去连翘外其余药材按原药方配比)。
2.3 专属性试验 精密吸取“2.2.3”项下的阴性样品溶液10μL,按“2.1”项下的色谱条件试验测定。结果表明,阴性样品对连翘酯苷A的含量测定结果无明显干扰(图1)。
2.4 线性范围 取连翘酯苷A对照品约5 mg,精密称定,置50 mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀(含连翘酯苷A 0.1 mg/mL),吸取上述溶液1 μL、2 μL、4 μL、10 μL、12 μL、14 μL分别进样,按“2.1”项下的色谱条件測定,以对照品量(μg)为横坐标,峰面积为纵坐标,绘制标准曲线,得到线性回归方程:Y= 2302379X + 2377,r=0.9999(n=6)。结果显示连翘酯苷A在0.1~1.4μg范围内呈现较好的线性关系(图2)。
2.5 精密度试验 按“2.2.2”项下对照品溶液制备方法配置对照品溶液,重复进样5次。结果显示连翘酯苷A对照品峰面积的RSD为0.40%(n=5),表明精密度良好。
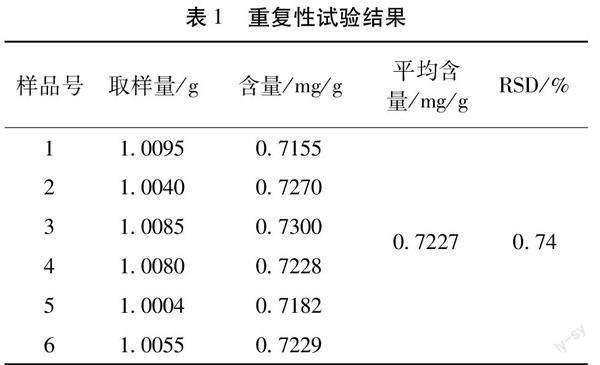
2.6 重复性试验 取相同批次样品(批号:20180514)6份,按“2.2.1”项下供试品溶液制备方法,制备样品,按“2.1”项下的色谱条件试验,测定6份样品的含量,结果显示6批样品的平均含量为0.7227 mg/g,RSD为0.74%(n=6),表明重复性良好(表1)。
2.7 稳定性试验 精密吸取样品溶液10 μL,按“2.1”项下的色谱条件,分别于0 h、2 h、4 h、8 h、12 h进行测定,记录各个时间点的色谱峰面积,结果显示样品溶液中的连翘酯苷A在12h内峰面积基本保持稳定,RSD为0.80%(n=5),表明样品在12 h内稳定。
2.8 加样回收试验 取已知含量的样品(0.7227 mg/g)粉末9份,每份取约0.5 g,精密称定,分别置于具塞锥形瓶中,3个具塞锥形瓶中精密加入0.2120 mg/mL的连翘酯苷A对照品溶液0.85 mL(约相当于供试品含量50%)、70%甲醇25 mL,另3个具塞锥形瓶中加入上述对照品溶液1.7 mL(约相当于供试品含量100%)、70%甲醇25 mL,剩余3个具塞锥形瓶中加入上述对照品溶液2.4 mL(约相当于供试品含量150%)、70%甲醇25 mL,按“2.1”项下的色谱条件试验,分别记录色谱图,计算平均回收率为93.34%,RSD为1.26%(表2)。
2.9 样品含量测定 取甘露解毒丸样品,按“2.1”项下的色谱条件试验并测定含量,结果显示2批样品平均含量为0.7770 mg/g,RSD为0.93%(表3)。
3 讨论
3.1 选择流动相 参照《中国药典》2020年版一部“连翘”项下的连翘酯苷A含量测定方法[8],以乙腈-0.4%冰醋酸溶液(15∶85)为流动相,进行实验分析,供试品中连翘酯苷A与其他成分达到较好的分离,理论塔板数较高,保留时间适宜,故作为检测流动相。
3.2 流速的选择 考察提取条件时,选择1.0 mL/min的流速,随着实验的进行,虽然持续清洗色谱柱,柱压仍偏高,故考察了0.8 mL/min流速,柱压相对理想,且含量无明显差异,故本次实验流速选择为0.8 mL/min。
3.3 检测波长的选择 通过考察连翘酯苷A的紫外光谱得出其在330 nm处有最大吸收,故选择330 nm作为检测波长。
3.4 提取效率的考察 试验分别考察了以70%甲醇作为提取溶剂,20 min、30 min、40 min超声处理提取。结果显示超声时间为30 min时,供试品中连翘酯苷A的含量保持相对稳定,故将提取时间设定为30 min。
3.5 提取溶剂的考察 以“3.4”项下选定的30 min作为提取溶剂超声提取时间(功率250 W,频率40 kHz),试验考察了25%甲醇、50%甲醇、70%甲醇3种不同提取溶剂对提取效率的影响。结果表明用70%甲醇作为提取溶剂时,样品中连翘酯苷A的含量最高,故将提取溶剂确定为70%甲醇。
3.6 色谱条件耐用性考察 采用SHISEIDO CAPCELL PAK C18(250 mm×4.6 mm,5 μm)和SHIMADZU Wonda Cract ODS-2 C18两种不同厂家的色谱柱(250 mm×4.6 mm,5 μm),按“2.1”项下的色谱条件分别试验,结果表明两种不同厂家色谱柱分离效果良好,含量测定结果无明显差别,表明耐用性良好。
当前,蒙医药事业的发展受到全社会的广泛关注,蒙药制剂源于与疾患斗争经验的药方,有着悠久的历史、丰饶的资源和鲜明的民族特征,其药效显著、使用广泛、受众者广。近年来,有关蒙药制剂的研究发展速度较快,目前已经运用色谱分离、波谱分析等方法对其中所含的化学成分进行广泛而深入地研究[9-11]。与此同时,蒙药制剂现行质量标准存在着质量控制指标单一、药效关联性不强等问题[12-13]。试验首次建立了高效液相色谱法测定甘露解毒丸中连翘酯苷A的含量,方法便捷科学、准确稳定,为建立相应制剂的质量标准提供了依据。
参考文献
[1]内蒙古自治区药品监督管理局.内蒙古蒙药制剂规范注释(第三册)[M].赤峰:内蒙古科学技术出版社,2022:126.
[2]佩媛,韩立柱,汪芸兰,等.连翘的研究进展及质量标志物的预测分析[J].中华中医药学刊,2022,40(4):19-27.
[3]陈日来,李玉珍,李衡梅,等.高效液相色谱法同时测定双黄连口服液中连翘酯苷A和连翘苷[J].中国医院药学杂志,2007,18(3):420-421.
[4]苏平菊,陈红燕,安青松,等.高效液相色谱法测定连花清瘟胶囊中连翘酯苷A的含量[J].中国现代药物应用,2014,8(14):251-252.
[5]黃如玉,唐瑞欣,厉言,等.UPLC-MS/MS法同时测定连花清瘟胶囊中11种成分[J].中成药,2023,45(1):24-28.
[6]蔡清宇,唐慧慧,李曼玲,等.高效液相色谱法测定连花清瘟颗粒中连翘酯苷A含量[J].中国中医药药信息杂志,2016,23(2):98-100.
[7]侯爱荣,李俊强.HPLC法测定黄连上清片中连翘酯苷A的含量[J].药学研究,2018,37(6):325-327.
[8]国家药典委员会.中华人民共和国药典(一部)[S].北京:中国中医药科技出版社,2020:177.
[9]白凤,李斌鑫,董玉,等.蒙药复方协日嘎四味化学成分的研究(Ⅱ)[J].内蒙古大学学报(自然科学版),2018,49(4):391-395.
[10]田华,邓雁如,周坤,等.蒙古黄芪的化学成分研究[J].中国实验方剂学杂志,2016,22(7):70-73.
[11]刘晓明,姜勇,孙永强,等.肉苁蓉化学成分研究[J].中国药学杂志,2011,46(14):1053-1058.
[12]佟海英,魏立新,奥·乌力吉,等.蒙药标准化的现状和存在问题及对策[J].医药导报,2019,38(4):452-455.
[13]朱小玲.蒙药的发展历史与研究进展[J].中华中医药杂志(原中国医药学报),2021,36(2):667-670.
(收稿日期:2023-05-17 编辑:陶希睿)
作者简介:樛跃(1986—),女,汉族,硕士,工程师,研究方向为生物化学与分子生物学。E-mail:1221sep@163.com
