5例伴单克隆免疫球蛋白沉积的增生性肾小球肾炎临床及病理分析
2024-03-01汪凤刘小妹徐佳奕王福妹苏雪松周华王艳秋
汪凤 刘小妹 徐佳奕 王福妹 苏雪松 周华 王艳秋
伴单克隆免疫球蛋白沉积的增生性肾小球肾炎(PGNMID)是一种具有肾脏意义的单克隆丙种球蛋白病,中老年常见,主要临床表现为蛋白尿、血尿及肾功能不全[1],肾脏组织病理改变主要为膜增生性肾小球肾炎(MPGN)[2];免疫荧光表现为单个免疫球蛋白G重链亚类和单个轻链亚型沉积;电镜下见系膜区、内皮下及上皮下颗粒状电子致密物[2];患者血液中可检测到异常克隆的浆细胞,但检出率低,常低于30%[3],易被漏诊和误诊。PGNMID治疗依赖于靶向可识别的B细胞或浆细胞克隆[4],但具体方案尚不清楚,约20%患者进展至终末期肾病[5]。PGNMID的病因及发病机制尚不明确,除血液系统恶性肿瘤外[6],还与自身免疫性疾病[7]、感染[8]、恶性肿瘤[9]等疾病相关。PGNMID早期诊断是改善预后的关键,因此,我们通过回顾性分析5例PGNMID患者的临床及病理特征,旨在提高对PGNMID的认识,减少漏诊和误诊。
对象与方法
1.对象:2018年1月~2022年1月于我院行肾脏活检并符合PGNMID诊断的患者5例。PGNMID的诊断标准[2]:(1)光镜:膜增生性或毛细血管内增生性肾小球肾炎或非典型膜性肾小球肾炎;(2)免疫荧光:单一免疫球蛋白G亚类和单个轻链亚型阳性,通常为免疫球蛋白G3和κ;(3)电镜:系膜、内皮下和(或)上皮下见电子致密物;(4)临床:缺乏冷球蛋白血症的临床表现或实验室证据。排除标准:血液系统恶性肿瘤、自身免疫性疾病、感染及恶性肿瘤。本研究已通过中国医科大学附属盛京医院伦理委员会审核批准,符合《赫尔辛基宣言》,因回顾性研究申请免知情同意。
2.方法
(1)资料收集:收集所有患者的人口统计学资料(包括性别、年龄)和临床资料[包括病程、高血压病史、临床表现、24 h尿蛋白定量(UTP)、尿红细胞计数(URBC)、血清白蛋白(ALB)、血清肌酐(SCr)、估算的肾小球滤过率(eGFR)、血红蛋白(Hb)、血清补体C3、免疫球蛋白定量、免疫固定电泳(IFE)结果、免疫球蛋白轻链和游离轻链测定结果、肾脏病理和骨髓穿刺检查结果等]。镜下血尿:离心后尿沉渣显微镜检每高倍镜视野有红细胞3个以上;肾病综合征:UTP>3.5 g和ALB<30 g/L;肾功能异常:SCr>104 μmol/L(男)或>84 μmol/L(女);低补体C3血症:血清补体C3<0.6 g/L。
(2)结局定义:完全缓解:尿蛋白<0.5 g/d和肾功能正常;部分缓解:尿蛋白较基线减少>50%或肾功能稳定时尿蛋白<2 g/d(SCr增加<20%);未缓解:不符合完全或部分缓解标准但未达到终末期肾病,包括持续性尿蛋白或进行性慢性肾脏病;终末期肾病:需要肾脏替代治疗[10]。
(3)肾脏病理检查:在超声引导下行经皮肾脏穿刺术,对穿刺组织行光镜、免疫荧光及电镜检查。肾脏穿刺组织制备HE、过碘酸雪夫(PAS)、基底膜六胺银(PASM)、Masson染色切片置于光镜下观察;多克隆兔抗人免疫球蛋白G、免疫球蛋白A、免疫球蛋白M、C3、C1q、κ及λ抗血清对标本染色,根据免疫荧光强度按“+”~“+++”分类并记录沉积强度及部位;肾脏穿刺组织经2.5%戊二甲醛固定和甲胺苯蓝染色后,置于透射电镜下评估。
3.统计学处理:计量资料以实际值表示,计数资料以例数表述。
结 果
1.一般临床资料:5例PGNMID患者中,男4例,女1例,年龄47~68岁,病程2.5~60.0个月。所有患者均有高血压病史,临床上均表现为肾病综合征,镜下血尿和肾功能异常多见,此外,轻度贫血也多见,但低补体C3血症少见,所有患者免疫球蛋白定量均正常,血清和尿液IFE均未检测到单克隆免疫球蛋白。PGNMID患者免疫学检查(包括IFE、免疫球蛋白游离轻链测定及游离轻链比率测定等)和血液学检查(包括骨髓免疫分型检查和流式细胞仪检查等)阳性率低(20.0%),仅病例5血清学检测到与免疫荧光下同型的κ轻链单克隆性;骨髓穿刺术浆细胞比值升高少见(20.0%),仅病例3骨髓中浆细胞比值升高,占骨髓所有细胞的3.0%;流式细胞术显示克隆的浆细胞占全部有核细胞的0.24%。所有患者均无淋巴结肿大、关节痛、皮肤溃疡等,风湿3项、抗核抗体、乙肝和丙肝检查均正常。见表1。
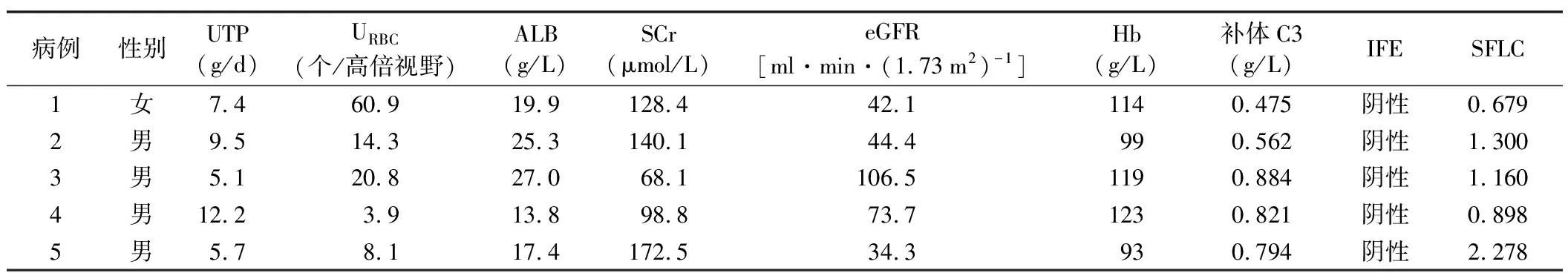
表1 5例患者的临床资料
2.肾脏病理资料:5例患者肾脏组织光镜下均呈MPGN样病变,其特征是病变的肾小球弥漫性分布,系膜细胞和基质弥漫性中至重度增生,向毛细血管壁广泛插入,导致毛细血管壁弥漫增厚,管腔狭窄。PASM染色可见基底膜呈双轨征或多轨征;Masson染色可见系膜区和内皮下嗜复红蛋白沉积。病变后期,系膜基质弥漫重度增生,毛细血管腔闭塞,肾小球毛细血管襻呈分叶状。见图1。免疫荧光检查见5例患者免疫球蛋白G3和κ轻链蛋白呈颗粒状和(或)团块状弥漫分

图1 PGNMID患者肾脏组织光镜检查结果[A:肾小球系膜区增宽,如箭头所示(HE染色,×400);B:肾小球系膜细胞和基质重度增生,毛细血管襻呈分叶状,如箭头所示(PAS染色,×400);C:肾小球基底膜弥漫增厚,系膜基质插入,可见“双轨征”,如箭头所示(PASM染色,×400);D:肾小球球囊周围可见纤维化,见小细胞性新月体形成,如箭头所示(PASM染色,×400);E:肾小球系膜区、内皮下可见嗜复红蛋白沉积,如箭头所示(Masson染色,×400);F:肾小球系膜细胞和基质重度增生,系膜区、内皮下可见嗜复红蛋白沉积,如箭头所示(Masson染色,×400)]
布于肾小球毛细血管襻和(或)系膜区;所有患者均伴补体C3和C1q沉积,均未见λ轻链蛋白、免疫球蛋白M和免疫球蛋白A沉积。见图2。电镜下5例患者电子致密物呈颗粒状或团块状沉积于肾小球,多见于系膜区、内皮下,少见于上皮下及基底膜内,均未见纤维丝样、微管样及结晶体样等亚结构。见图3。5例患者肾脏组织病理检查结果见表2。

图2 PGNMID患者免疫荧光检查结果[A:免疫球蛋白G颗粒状沉积于肾小球系膜区及毛细血管襻(×400);B:单克隆免疫球蛋白G3颗粒状沉积于肾小球毛细血管襻,呈“花瓣状”(×400);C:C3颗粒状沉积于肾小球系膜区及毛细血管襻(×400);D:C1q颗粒状沉积于肾小球毛细血管襻,呈“花瓣状”(×400);E:κ轻链蛋白颗粒状沉积于肾小球毛细血管襻,呈“花瓣状”(×400);F:λ轻链蛋白染色阴性(×400)]

图3 PGNMID患者电镜检查结果[A:肾小球上皮细胞足突弥漫融合,内皮下(如红色箭头所示)及系膜区(如蓝色箭头所示)可见大量高密度电子致密物沉积(×5 000);B:系膜区可见大量高密度电子致密物沉积(如蓝色箭头所示,×5 000)]

表2 5例患者肾脏病理组织检查结果
3.临床结局:病例5为新诊断患者,现BCD(B硼替佐米,C环磷酰胺,D地塞米松)方案治疗中,无随访资料,余4例患者随访均超过12个月,随访中血清及尿液IFE持续阴性,骨髓学复查浆细胞比值较前下降,支持治疗预后差,随访3年后均进展至终末期肾病,免疫抑制剂治疗均可获得临床缓解。见表5。

表5 4例患者临床结局
讨 论
PGNMID是一种具有肾脏损伤的罕见的单克隆丙种球蛋白病,占肾脏活检的0.17%~3.70%[5],本研究分析了5例PGNMID患者的临床及病理特征以引起人们对其认识及重视。
PGNMID临床表现无特异性,国内外研究结果均显示其肾脏损害以大量蛋白尿和低蛋白血症为主,肾功能不全及镜下血尿常见[10-11],本研究中的患者也以肾病综合征突出,肾功能异常、镜下血尿多见。与国内研究[11]结果一致,本研究中的PGNMID患者也以中老年男性常见,高血压发生率高,但补体C3下降少见,轻度贫血常见,且为正细胞正色素性。
肾脏病理学评估对PGNMID诊断必不可少,病理改变主要集中于肾小球,光镜下常表现为MPGN样病变,病变明显病例可见新月体形成,以细胞性新月体多见。与既往文献[2,5,10]报道一致,本研究中的5例患者光镜下为MPGN样病变,小细胞性新月体少见,但1例患者伴结节性病变,我们排除了糖尿病肾病,这在其他文献中也见报道[12]。既往研究报道PGNMID光镜下可表现为毛细血管内增生性肾小球肾炎、膜性肾小球肾炎和系膜增生性肾小球肾炎[10],但在本研究中均未发现,需增加病例数进一步探讨。免疫荧光下肾小球免疫球蛋白和轻链沉积的限制性是诊断PGNMID的基石,与既往研究报道一致[10,13],本研究中患者肾小球见免疫球蛋白G3和κ轻链沉积,免疫荧光强度高,这可能是因为免疫球蛋白G3分子量大难以透过肾小球滤过膜,能通过其Fc结构域与细胞膜上的Fc受体相结合,选择性积聚在肾小球[10],并通过局部和全身作用于经典途径,激活下游炎症介质促进肾小球白细胞浸润和增生,导致肾小球肾炎[14]。与既往文献报道一致[5,10],本研究中PGNMID患者电子致密物颗粒状和(或)团块状沉积于肾小球,未观察到类似纤维丝、微管、结晶体样等亚结构,沉积物多见于内皮下和系膜区,少见于基底膜内和上皮下。肾脏病理检查提示PGNMID时需免疫学和血液学检查以确定是否存在病理性浆细胞疾病[2,5]。免疫固定电泳检查因可实现单克隆免疫球蛋白的鉴定及分型,常作为首选检查[15],而血清免疫球蛋白游离轻链测定敏感度高,SFLC可提示轻链亚型的克隆性[16]。Nasr等[10]报道约30%患者存在异常蛋白血症,但本研究中患者血清或尿液IFE未见单克隆蛋白,1例SFLC异常,且表现为与免疫荧光下相同的κ轻链单克隆性。骨髓学检查浆细胞克隆阳性率低,本研究中仅1例患者骨髓液流式细胞术见浆细胞克隆,克隆检出率与Bhutani等[3]的报道相近。鉴定致病单克隆免疫球蛋白对PGNMID的诊断和预后具有重要意义,免疫固定电泳、免疫球蛋白游离轻链测定及骨髓检查互相补充,能提高免疫球蛋白阳性率,从而发现PGNMID致病因素。
PGNMID具体治疗方案尚不清楚,目前认为靶向克隆[4]和免疫抑制剂[10,17]治疗能改善肾脏预后。本研究中的患者采用免疫抑制剂治疗后可获得临床缓解。病例4病史长达5年,既往采用免疫抑制剂治疗,确诊后继续激素及环磷酰胺治疗,1年后完全缓解,表明积极干预治疗可改善患者预后。PGNMID预后异质性大[10],约20%患者在诊断后2~5年进入终末期肾病,本研究中1例患者在随访3年后进展至终末期肾病,最终行肾脏替代治疗,但本研究患者少且部分患者随访时间短,需要进行更大规模的研究和更长时间的随访,从而确定疾病的性质,制定最佳治疗方法,以研究疾病的进程。
PGNMID是一种罕见的单克隆免疫球蛋白病,其病因及病理生理机制不明确,临床表现和实验室检查无特异性,疾病诊断依赖于肾脏活检,因此临床表现为蛋白尿、血尿或肾功能异常的患者应尽早行肾脏病理检查,以减少PGNMID的漏诊和误诊。
