Bardet Biedl 综合征的诊治进展
2024-02-25徐鑫星王传凯蒋丽琼综述王春林审校
林 娇 徐鑫星 王传凯 蒋丽琼综述 王春林审校
浙江大学医学院附属第一医院儿科(浙江杭州 311100)
Bardet-Biedl综合征(Bardet Biedl syndrome,BBS,OMIM 209900)最初是由Bardet于1920年,随后Biedl于1922年报道的一类遗传病,迄今已发现26种基因变异类型,且这26种基因都定位于初级纤毛上,因此也被认为是一种纤毛病。BBS 的发病率存在地区差异,中东相对常见,发病率约为1∶135000,欧洲和北美约为1∶160000~1∶140000[1]。中国尚无明确统计数据,查阅国内外各数据库显示发病例数约为百例,足可见其罕见性,且目前缺少对该疾病系统的综述。因此,本文从BBS的临床特征、诊断标准、致病基因、基因与表型相关性、我国BBS患者现状、治疗等方面进行综述,希望为BBS临床诊治提供一些价值。
1 临床表现及诊断标准
BBS 临床表现多样,主要特征如下:①视网膜病变。研究表明90%的患者会出现由视锥、视杆细胞进行性萎缩引起的视网膜病变。病初患者多表现为夜盲症,继而出现进行性周围性视力丧失、色差辨识度下降,最后视力全部丧失,98%的患者在30 岁前完全失明[2]。同时此类患者在起病前2 年视网膜电位就会有所衰减,因此视网膜电图可以作为早期监测的指标。②多指(趾)畸形。2/3的患者会出现多指(趾)畸形,多为轴后多指,多趾畸形多见多指,部分患者会同时合并有短指或并指畸形。③肥胖。多为向心性肥胖,出生时患者的体重通常在正常范围内,但接近正常上限,到5岁时90%以上的患儿会发展为超重或肥胖[3]。Moore等[4]一项长达22年的前瞻性研究表明,随着时间推移 BBS患者都会出现肥胖,25%的患者甚至达到病态肥胖的程度(BMI>40 kg/m2)。同时,肥胖也会增加BBS患者心血管、代谢综合征等疾病的发生率。④性腺发育不良。发生率约在59%,男性的发生率比女性更高,常报道有小阴茎、小睾丸、隐睾及低促性腺激素性性腺发育不良等现象。部分男性患者存在生殖道囊肿导致的生殖道部分阻塞,从而导致生育功能低下。女性患者大多在青春期时才有表现,例如月经周期不规律或多囊卵巢,也可能存在复杂的泌尿生殖畸形如阴道闭锁、膀胱阴道瘘、双子宫等[5];⑤智力低下:发生率约50%~61%,表现为发育迟缓、语言障碍、行为异常等,磁共振成像可显示海马灰质、右下纵束白质体积减少[6]。Kerr等[7]对24例患者进行了神经发育评估,发现BBS 患者的智力比平均值低1.5 个标准差,但只有20%~25%患者符合智力障碍的诊断标准,大多数患者为轻度的认知障碍。⑥肾功能异常。肾脏病变是BBS患者的主要特征,同时也是导致死亡的主要病因。然而BBS患者在肾脏方面的临床症状通常不典型,约1/3的患者在儿童时期会出现尿液浓缩功能下降,表现为多饮多尿,烦渴,夜尿多等。如果病初多饮多尿的症状未被重视,可能导致确诊时已进展至终末期肾病(end stage renal disease,ESRD)[8]。据报道BBS肾脏累及率大约在 53%~82%。近期一项英国研究纳入了350 名BBS 的患者发现,31%的儿童和42%的成人存在慢性肾脏病,其中6%的儿童和8%的成人患者已进入ESRD,且所有ESRD儿童患者的起病年龄均在5岁之前[9]。此外,BBS患者常伴有肾脏结构的异常,肾囊肿最为常见,其他还表现为肾发育不全,皮髓质分界不清,马蹄肾等。B 超检查在出生时或胎儿期可发现肾脏外形增大,出现单发或多发囊性病变,或肾脏弥漫均匀的高回声,无任何皮髓质的分化。因此BBS 患者的管理指南建议每位患者进行基线肾脏超声检查,以检测是否存在结构异常。
BBS 的次要特征还包括先天性心脏病,肝脏受累(肝脏纤维化、肝囊肿等),内分泌疾病如糖尿病、高胆固醇血症等,牙釉质发育不良,共济失调,言语、生长发育迟缓,颅面部畸形,嗅觉异常等。其中胰岛素抵抗较为常见,然而2型糖尿病的发生率并不高。据文献统计[10],15%患者在33岁时会进展为糖尿病,到43岁时糖尿病的发生率为48%。口腔和牙异常也较常见,患病率>50%,可表现为:高弓腭、咬合不正、牙齿拥挤、牙根短等。50%的BBS患者存在嗅觉减退,但由于临床中难以对其进行准确的评估,该百分比可能会更高。此外,心血管病是BBS 中观察到的另一种次要特征,Niederlova 等[11]曾报道心脏异常的发生率为29.8%。BBS的临床诊断标准为满足6项主要标准中4项或3项主要标准加2项次要标准即可[6]。但是BBS 是一种高度异质性的疾病,首先并非 BBS的所有临床症状都会发生在单个患者身上,即使是同一个致病基因,其临床表型也并不一致,其次有些特征的出现也与患者的年龄有关,这些情况的存在均有可能延误诊断[12]。因此,当存在BBS 中2 种表型时,建议多学科临床评估患者是否需要进行全外显子检测或全基因组检测明确BBS的遗传基因[13],这样更有助于早期识别及管理疾病。
2 BBS与相关纤毛病
目前已发现26 种BBS 的致病基因都定位于初级纤毛,参与了纤毛的功能和构成。纤毛是一种以微管为主要结构突出于细胞表面的线性细胞器,存在于人体几乎每种细胞的表面。按超微结构可将其分为两大类:动力纤毛和初级纤毛。初级纤毛存在于大多数细胞的表面,包括肾脏、胆管、胰脏的上皮细胞等,在细胞增殖、迁移、分化与细胞周期的调控等生理活动中起关键作用[14]。因此,纤毛组装和功能的缺陷会导致多种先天性疾病,例如BBS、Joubert综合征、常染色体显性多囊肾病、常染色体隐性多囊肾病和Meckel 综合征等。纤毛病的特点是涉及核心组织或器官如视网膜、大脑和肾脏等,使得在纤毛病及相关疾病中可观察到广泛和重叠的临床特征,如:肾功能异常,肾囊肿形成,视网膜变形,先天性肝纤维化、性腺功能异常等[15]。BBS 致病基因均参与纤毛的构成,因此也属于纤毛病的范畴。BBS 基因变异可分为四类,大多数是由于编码BBSome 的基因(BBS1,2,4,5,7,8,9,17,18)发生变异。第二大类是编码辅助BBSome组装的伴侣蛋白基因变异(BBS6/MKKS,10,12)。第三类则是BBS3/ARL6变异,这是一种辅助BBSome功能的GTPase[16]。第四类中其他BBS 蛋白质在纤毛的基部或中心体独立发挥作用,招募BBSome 如BBS 14、BBS 15和BBS 16在中心粒随体上行使功能,促进BBSome的招募[1]。可见BBS发病与BBSome的功能障碍密切相关。BBSome是八聚体蛋白复合体,在大多数纤毛生物中具有高度进化保守性,主要参与蛋白质的运输过程,可将蛋白质运输到纤毛和质膜上[17]。而这些BBS蛋白的变异会导致关键分子定位错误,破坏下游关键的细胞信号通路,包括Sonic Hedgehog 信号通路、瘦素信号通路、光感受器信号通路和 G 蛋白偶联受体介导的信号通路[18],从而影响细胞周期调控、增殖和细胞平面极性等[19]。
3 BBS致病基因
迄今为止已发现多达26 种变异基因(BBS 1~BBS 21、IFT 74、SCLT 1、SCAPER和NPHP 1等),BBS1、BBS2和BBS10是最常见的变异基因,占总数的50%左右。
BBS 1基因位于第11 号染色体(11 q 13.2)的长臂上,其大小约为23kb,编码593个氨基酸组成的蛋白质产物,包括17个外显子。该基因在睾丸、视网膜、脂肪组织、心脏、骨骼和胰腺组织中均有表达,肾脏中表达最高。BBS 1是BBSome 复合体的重要元件,它可通过增加Rab 8-GTP 促进纤毛膜的合成,同时在瘦素受体信号通路中也起着一定作用。然而,有体外研究表明BBS1的缺失对 BBSome 的形成和稳定性影响很小[20]。p.M390R 是最常见的BBS1变异,约占欧洲人BBS1变异率的80%[21]。
BBS 2定位于16 号染色体16 q 13 区域,由18 个外显子组成,编码721个氨基酸。广泛表达于脑、肾、肾上腺和甲状腺等多种器官。BBS2作为BBSome核心复合体的一部分,促进初级纤毛膜的生物合成。研究指出,BBS2中的任何致病序列变异都会导致肥胖和视网膜病变[22]。
BBS 3/ARL 6基因位于3 号染色体(3 p 11.2)的短臂上,由9个外显子组成,编码由186个氨基酸组成的胞质蛋白,被称为ADP 核糖化因子样蛋白6。ARL6呈现组织特异性表达,在大脑和肾脏中的表达水平最高。ARL 6是小GTP 酶Arf 家族的成员,具有两亲性N端螺旋,可以通过GTP依赖性方式与膜结合,直接调控BBSome 进入纤毛[23]。
BBS6/MKKS位于20号染色体短臂(20p12.2),由6个外显子组成,编码570个氨基酸组成的蛋白质,是辅助BBSome组装的伴侣蛋白。BBS6 是一种起源中心体的蛋白,胞质分裂时也存在于中间体。该蛋白与伴侶素家族中第二家族蛋白起相似作用,参与其他蛋白的折叠和稳定过程,其失活可以导致其他蛋白的错误折叠[24]。目前报道的所有变异个体中,只发现杂合突变。
BBS10,染色体位置为12q21.2,由两个外显子组成,编码723个氨基酸组成的大分子蛋白,属于新型Ⅱ型伴侣蛋白亚家族,由三个功能域组成,包括赤道域、中间域和顶端域[25]。BBS10是辅助BBSome组装的伴侣蛋白,具有伴侣蛋白的结构域。目前发现BBS10与肥胖发生密切相关,其位于初级纤毛的基体内,降低其表达会抑制纤毛的生长,最终导致糖原合酶激酶3途径磷酸化增加并诱导过氧化物酶体增殖物激活,从而增加脂肪生成。此外,BBS10可直接与胰岛素受体相互作用从而参与胰岛素信号转导[24],Wang等[26]发现当BBS10变异时会破坏人成纤维细胞和iPSC分化的神经元中的胰岛素信号转导,同时下丘脑神经元中的瘦素信号转导也会受损。BBS 10变异频率较高,约为20%,是BBS变异的关键基因。
除以上特征基因外,BBS 与其他纤毛病之间存在重叠的基因,例如NPHP 1变异也能导致肾单位肾痨以及Joubert 综合征;BBS 13(MKS 1)变异与Joubert 综合征和Meckel-Gruber 综合征均有关联;BBS 14(CEP 290)基因变异能导致Leber 先天性黑蒙、Senior-Løken综合征、Joubert综合征和Meckel-Gruber综合征。而IFT172与Joubert综合征和短肋骨-多指综合征存在关联[27]。可见BBS基因具有多效性,同一基因变异能导致不同的疾病,因此在做基因诊断时,需要同时结合临床表型、遗传特性等。
4 BBS基因型与表型相关性
BBS变异基因较多,准确描述基因型-表型的关系需要较大样本量,这限制了在BBS此类罕见病中得出确切的结论,但根据既往研究报道也有迹可循。Niederlova等[11]在一项meta分析中将BBS患者的5项临床特征[视网膜病变、多指(趾)畸形、肥胖、认知障碍和肾损害]各列为0.2 分,总分为1 分,比较不同基因引起临床症状的差异性,结果发现BBS 3/ARL 6变异症状评分明显低于BBSome 或伴侣BBS基因变异的患者,说明BBS 3/ARL 6变异的患者通常比其他基因变异的患者存在更少的临床症状。在BBSome基因中,BBS1和BBS8/TTC8变异患者的平均评分最低,而BBS2和BBS7变异患者的平均症状评分最高。对于不同表型,其变异基因也有所不同,BBS 1基因引起的视网膜病变通常较轻,而BBS 2、BBS3和BBS4基因变异常表现出严重的眼部症状[5]。BBS 2变异的患者多指畸形率更高,BBS 10基因变异表现出更明显的肥胖和胰岛素抵抗[28]。对于肾脏病变,BBSome 的核心基因(BBS 2、BBS 7或BBS 9)肾脏病变发生率超过60%。Imhoff等[29]分析了一个由33例BBS患者组成的法国队列,发现含有BBS6、BBS10和BBS12基因变异的患者比其他成分缺陷的BBS 患者有更严重的肾脏疾病[8]。Meyer[30]统计了44例发生肾衰竭的患者,发现BBS10是最常见的基因。随着BBS 致病基因谱不断扩充和完善,今后会有更详细和全面的基因-表型数据库。
5 我国BBS现状
我国目前尚无BBS患病率相关数据,查阅截止至2022年9月在各中英文期刊上发表的关于中国人BBS 患者的文献,进行了病例汇总分析,共纳入73篇文献139例BBS患者,纳入患者年龄(16.1±9.7)岁,其中男性82例(59.0%),年龄(16.3±9.8)岁。在临床症状方面,91.4%(127/139)患者存在视网膜的病变,患有肥胖症及多指(趾)畸形的人数也较多,分别为125和124例,智力低下的发生率77.7%(108/139),与国外的发生率相比偏高,而性腺发育不良及肾功能异常的发生率相对偏低,分别为54.0%(75/139)和36.7%(51/139)。此外,我国BBS 患者其他常见的异常表现还包括先天性心脏病、糖代谢异常、口腔和牙异常等(表1)。利用二代测序技术,有77 例患者已明确遗传学诊断,最常见的变异基因包括BBS7、BBS2、BBS10及BBS9,详见表2。12 例患者为产检时发现问题。其中11 例表现为泌尿系统异常合并多指(趾)畸形,1 例患儿超声显示肾脏增大、回声增强。通过遗传学检查诊断,所有患者均为BBS患者[31-37]。这些研究涉及检测样本来源各不相同,7例患儿由羊水穿刺或绒毛穿刺取得标本,2例为流产后选取脐带组织和脐带血进行遗传学检测,另有3例是同一对父母所生,宫内超声均表现为泌尿系统异常、多指畸形合并脑室增宽,但该3例患儿未留取到样本,仅通过对父母进行全外显子测序发现父母均携带BBS1变异[31]。以上12例患儿常见基因变异类型为BBS7(4/12,33.3%)和BBS1(5/12,41.7%)。
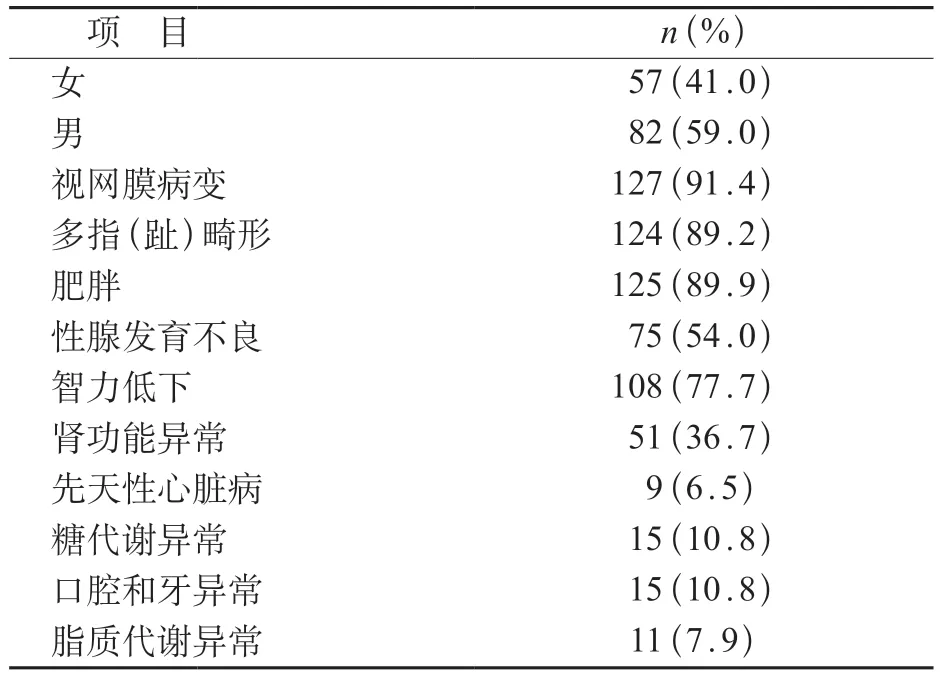
表1 我国Bardet Biedl综合征临床症状人数统计(n=139)

表2 我国Bardet Biedl综合征致病基因统计(n=77)
综上所述,目前BBS仍存在确诊年龄偏大现象,但近几年随着基因检测技术的进步以及对该疾病逐渐熟悉,临床医生能更早识别疾病,甚至在产前做出诊断,对提高我国儿童素质与健康,降低家庭负担具有重要意义。
6 治疗
BBS 目前没有治愈性疗法,治疗旨在预防或治疗相应的症状和并发症,包括监测和控制高血压、糖尿病、肥胖症和肾功能衰竭等。多指畸形可通过手术矫正。对于视网膜病变,目前无治疗方法阻止其恶化,只能通过使用视力辅助设备和行动训练,使患者逐渐适应视力丧失。肥胖患者可通过低热量和蛋白质饮食、运动等方法进行早期管理以免发生代谢综合征,尽量减少对已经受累的器官特别是眼睛和肾脏的二次打击。有部分肥胖患者选择减肥药物或者手术也有不错的效果,但无长期随访数据,无法详知其后遗症及复发情况。对于肾脏畸形和肾功能异常的患者,必须密切监测肾功能,如果发展至ESRD,可选择肾透析或肾移植。随着对BBS基因及其致病机制的研究,也为治疗开阔了新的视角。Seo和Simons 等[38-39]通过向实验小鼠视网膜下注射含BBS腺病毒载体,可纠正视紫红质的错位,改善小鼠视网膜电位。Cring等[40]将人类野生型BBS1基因杂交到Bbs1M390R/M390R小鼠上,发现杂交后的雄性小鼠能够繁殖后代。此外还有特异性药物靶向治疗手段,赛美拉肽是一种MC 4 R 激动剂,可恢复MC 4 R 通路的活性,2022年已被FDA批准用于6岁以上BBS肥胖患者。Robert 等[41]对10 例BBS 患者进行为期1 年的2期临床试验,皮下注射赛美拉肽肥胖型患者体重较基线下降约16%。3 期临床试验也再次证明了赛美拉肽的有效性,总共纳入32 例患者,32.3%的患者在52 周时体重下降超过10%,同时饥饿感、腰围及脂代谢指标均有下降。副作用方面,主要是注射部位红肿以及皮肤色素沉着,并无严重并发症[42]。对于肾脏方向,在BBS斑马鱼模型中抑制特定信号分子,例如用雷帕霉素阻断mTOR 蛋白或用塞利西利布选择性阻断细胞周期蛋白依赖性激酶,可部分恢复肾脏的结构和功能[43]。上述一些治疗仍处于研究状态,也为今后更精准化的治疗提供了方向。
7 总结
BBS 临床表型和基因型多样,过去对于此类累及多系统的疾病了解不深,常不能给出确切的结论,但随着基因检测技术的进步,特别是对于具有广泛表型谱的遗传性疾病,二代测序能在一次测试中以相对较低的成本分析所有基因。可以帮助早期诊断,避免对患者进行不必要的诊断操作,如肾脏或肝脏活检,明确当前及以后的治疗,方便评估疾病的预后,同时可以强调未来可能出现的并发症,有助于重点筛查和更好的预防。综上所述,BBS 是一种较为复杂的疾病,其发病主要由于BBSome 相关基因变异,目前已发现26种致病基因。尽管 BBS 尚无治愈性疗法,但早期诊断对于改善生活质量、控制高血压、控制肥胖和防止肾功能进行性恶化非常重要。因此BBS 患者需要多学科共同管理,以获得更高的生存率及生活质量。
