海南省少数民族地区脂肪酸氧化代谢病新生儿筛查结果分析
2024-02-25黄慈丹许海珠温英梅刘秀莲
黄慈丹 许海珠 温英梅 刘秀莲
海南省妇女儿童医学中心 海南省新生儿疾病筛查中心(海南海口 570206)
脂肪酸β 氧化代谢障碍(fatty acid oxidative metabolism disorder,FAOD)是由于脂肪酸进入线粒体进行β 氧化代谢途径中的酶或转运蛋白功能缺陷,导致脂肪酸β 氧化代谢发生障碍引起的一组疾病。FAOD 包括原发性肉碱缺乏症(primary carnitine deficiency,PCD),短链酰基辅酶A脱氢酶缺乏症(SCADD)、中链酰基辅酶A 脱氢酶缺乏症(MCADD)、极长酰基辅酶A 脱氢酶缺乏症(VLCADD)、肉碱棕榈酰转移酶缺乏症Ⅰ(CPTⅠ)、肉碱棕榈酰转移酶缺乏症Ⅱ(CPTⅡ)、肉碱酰基肉碱移位酶缺乏症,β-酮硫解酶缺乏症及三功能蛋白缺乏症等,均属于常染色体隐性遗传病[1]。遗传代谢病据报道在新生儿人群中发病率高达0.5%[2]。串联质谱检测新生儿干血斑中的氨基酸谱与酰基肉碱谱,可以实现氨基酸代谢病、有机酸代谢病和脂肪酸代谢病共40余种疾病的群体筛查。随着大量遗传代谢病患者的被发现[3-4],已刷新了人们对此类疾病的认知,如PCD在法罗群岛发病率高达1∶30[5]。据报道海南省少数民族地区[6]和黎族人群[7]的PCD发病率较高,分别为1/3740及1/2295。本文总结海南省少数民族地区新生儿筛查中心串联质谱筛查资料,分析海南省少数民族地区FAOD 发病率及确诊患者的临床特点。
1 对象与方法
1.1 研究对象
收集2016年10月至2021年12月海南省内少数民族自治市县(琼中黎族苗族自治县、保亭黎族苗族自治县、陵水黎族自治县、乐东黎族自治县、昌江黎族自治县、白沙黎族苗族自治县和五指山市)出生满72 小时的共66 578 名新生儿(汉族29 043 名、黎族34 285名、苗族1 903名、其他民族1 347名)的数据。遵循知情同意自愿原则采集足跟血制成新生儿筛查干血斑样本46 285名(汉族18 299名、黎族25 713名、苗族1 313名、其他民族960名,男性25 114名、女性21 171名),参与率达69.5%。
样本采集严格遵循《新生儿疾病筛查技术规范(2010 年版)》要求。本研究获医学伦理委员会批准(HNWCMC伦审2022年第[35]号)。
1.2 方法
1.2.1 串联质谱筛查 以新生儿干血斑为样本,利用串联质谱(TQD型串联质谱仪,美国Waters公司)法分析干血斑中游离肉碱(free carnitine,C0)及酰基肉碱谱水平。本实验严格按照非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒(串联质谱法)操作说明书和液相串联质谱仪操作规程进行。
1.2.2 尿气相质谱有机酸检测 收集疑似FAOD的新生儿新鲜尿液,利用尿气相质谱法(7890B型气相质谱仪,美国Agilent 公司)分析尿中二羧酸类等脂肪酸代谢途径中间产物或终产物水平。本实验严格按照尿有机酸分析配套试剂盒操作说明书和气相质谱仪操作规程进行。
1.2.3 基因变异检测 采集疑似FAOD 的新生儿及其父母双亲外周血各2 mL,制成EDTA抗凝血,外送重庆迈基诺基因科技股份有限公司,进行FAOD相关基因的测序验证。
1.3 统计学分析
采用SPSS 17.0 统计软件进行数据分析。计数资料以例数(百分比)表示,组间比较采用χ2检验或Fisher精确概率法检验。以P<0.05为差异有统计学意义。
2 结果
2.1 海南省少数民族地区新生儿人群FAOD发病率
采用串联质谱共筛查新生儿46 285名,初筛阳性513 例(男282 例、女231 例),初筛阳性率1/90,召回501例(男286例,女215例),召回检测阳性31例(男17 例,女14 例),诊断FAOD 11 例(男7 例,女4例),发病率1/4208。男女间FAOD的初筛阳性率(χ2=0.95,P=0.331),召回阳性率(χ2=0.00,P=0.984)和发病率(χ2=0.08,P=0.784)差异均无统计学意义。11例确诊患儿的初筛游离肉碱及酰基肉碱检测结果见表1。
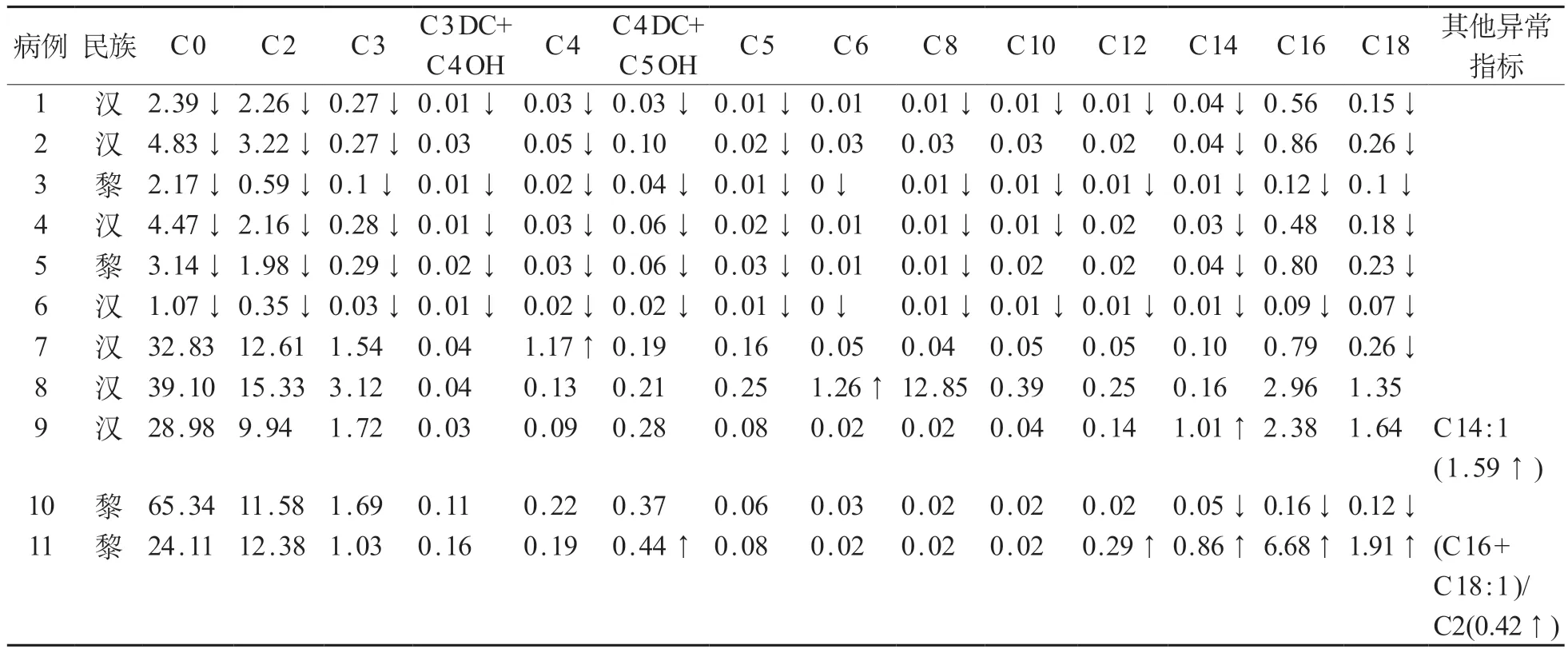
表1 11例确诊FAOD患儿初筛游离肉碱及酰基肉碱检测结果(µmol·L-1)
2.2 海南省少数民族地区新生儿人群FAOD 病种分布
诊断FAOD患儿11例中PCD 6例(汉族4例、黎族2例)、SCADD 1例(汉族)、MCADD 1例(汉族)、VLCADD 1例(汉族)、CPTⅠ 1例(黎族)、CPTⅡ 1例(黎族)。
2.3 筛查确诊FAOD患儿基因检测结果
11例FAOD患儿基因检测结果见表2。6例PCD患儿中携带纯合突变3 例,携带复合杂合突变3 例,共4 种突变,均为已报道突变。1 例SCADD 患儿检出ACADS基因c.889C>T和c.256G>A复合杂合突变。1例MCADD患儿检出ACADM基因c.1097G>A纯合突变。1 例VLCADD 患儿检出ACADVL基因c.664 G>A 和c.1819 T>G 复合杂合突变。1 例CPTⅠ患儿检出CPT1A基因c.1163+4C>T纯合突变。1 例CPT Ⅱ患儿检出CPT 2基因c.1408 G>T 和c.1102G>A复合杂合突变。

表2 11例FAOD病种分布及其基因检出情况
2.4 FAOD患儿随访结果
PCD 6 例中,男5 例、女1 例,均为足月儿;5 例确诊时均无临床症状,补充左卡尼汀后,血C0浓度均在正常范围,智力和语言发育正常;1例患儿初筛异常,多次通知家长拒绝召回,1岁时发现肝功能异常住院,未好转,住院期间发现心肌受损、致密化不全,射血分数低,心电图异常,经查C0浓度低,临床诊断为PCD,补充左卡尼汀,各项指标好转,体格和智力发育正常。SCADD患儿采用低脂饮食,适当补充肉碱或维生素B2,避免饥饿,随访智力发育、体格检查未见异常。MCADD 患儿确诊后采用低脂肪饮食,避免饥饿和疲劳,未用药物治疗。VLCADD患儿3岁5个月因感冒、纳差,出现低血糖昏迷,经输注葡萄糖后治疗后症状缓解。11 例FAOD 患儿的治疗随访情况见表3。
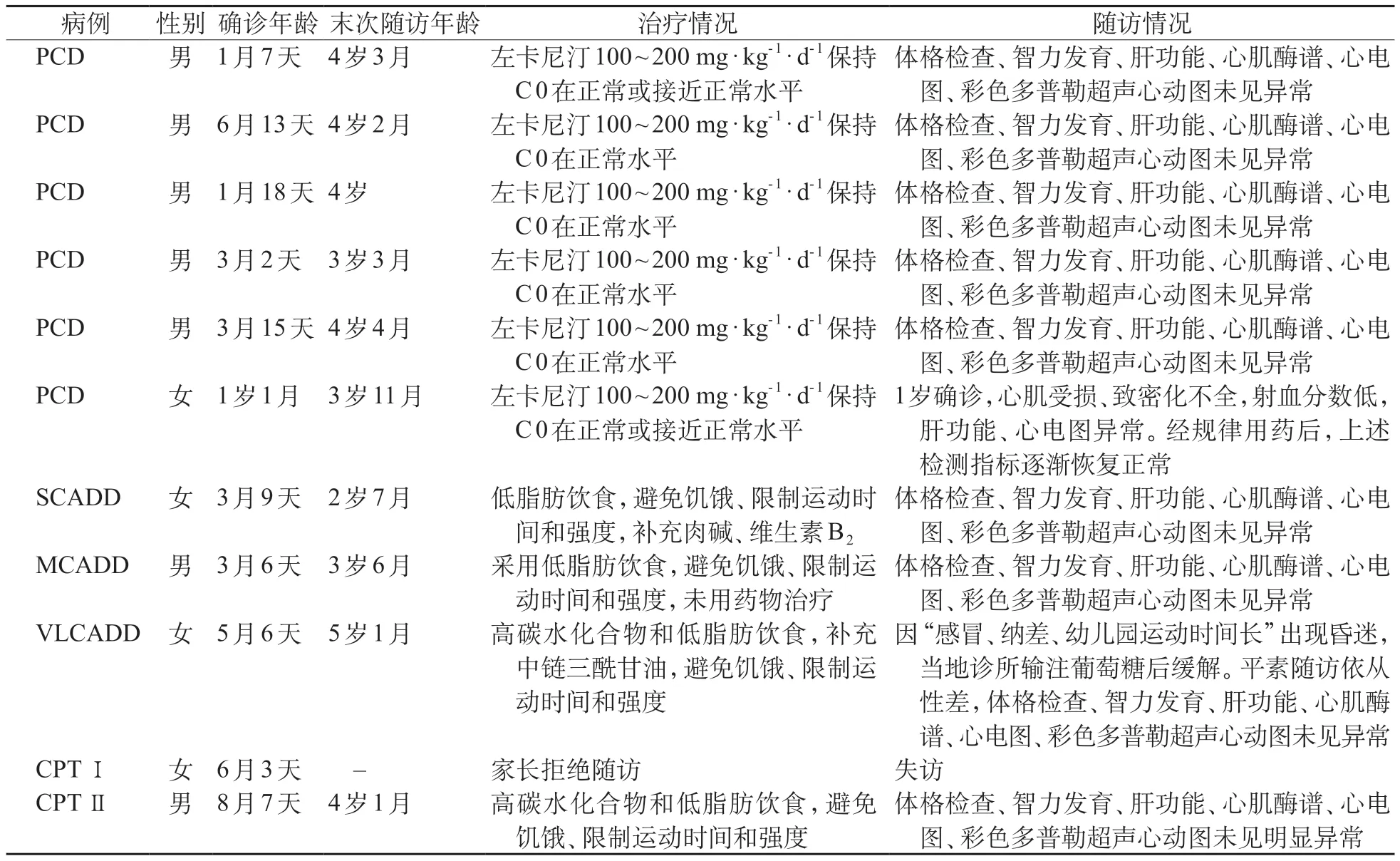
表3 11例FAOD患儿治疗随访情况
3 讨论
新生儿遗传代谢病是指有异常生化标志物的一大类疾病,绝大部分属于常染色体隐性遗传,少数为常染色体显性遗传,根据其异常的生化标志物分类为FAOD、氨基酸代谢病、有机酸尿症、碳水化合物代谢异常等[8]。目前串联质谱技术通过检测血氨基酸谱和酰基肉碱谱对氨基酸、有机酸及FAOD三类共40余种疾病进行筛查,已作为筛查遗传代谢病的常用技术[9]。筛查FAOD 类疾病约10 多种,其中PCD发病率较高[10-11],且PCD是FAOD疾病中预后较好的疾病,早期筛查意义重大。线粒体脂肪酸氧化属细胞能量的主要来源之一,禁食期间可提供80%的能量[12],空腹或长时间运动时,长链脂肪酸通过脂肪活化释放出脂酰辅酶A。长链脂肪酸不能通过线粒体内膜,需要肉碱协助进入线粒体,肉碱通过高亲和力的肉碱转运体转运至细胞[13]。在此过程中任何一个步骤发生障碍,即导致脂肪酸代谢受阻,乙酰辅酶A生成减少,ATP相应减少,能量合成障碍,引起相关疾病。
美国、澳大利亚、德国、意大利等国家FAOD的总体发病率为1/14545~1/6033[14-17]。本研究结果显示海南省少数民族地区FAOD 发病率为1/4208,高于上述国家的发病率,与国内报道的相比,高于浙江省的1/12261[18]。海南省少数民族地区新生儿FAOD 的发病率与国内先天性甲状腺功能低下症(CH)的平均发病率(1/3000~1/4000)[19]相近。CH早已被列入《新生儿疾病筛查管理办法》的必筛病种之一,鉴于海南省少数民族地区FAOD 发病率较高,建议把新生儿遗传代谢病串联质谱筛查列入海南省新生儿常规筛查项目,以提高海南省出生人口素质。
本研究11 例确诊病例中汉族7 例(发病率1/2614),黎族4 例(发病率1/6428),包含6 种疾病,PCD 患儿最多,与国内相关报道类似。PCD 是由于细胞膜上高亲和力的肉碱转运体肉碱转运蛋白基因变异所致的一种FAOD,是常染色体隐性遗传病,其发病率在在日本约1∶40000[20],在澳大利亚为1∶37000~1∶10000[21],PCD被认为是一种潜在致死性疾病[22]。肉碱在细胞内积累会降低脂肪酸的氧化,如果没有及时补充肉碱,PCD 症患者在生命早期就会出现急性代谢失代偿,或晚期出现骨骼和心肌病或心律失常致突然死亡[23]。此病在新生儿期偶有轻微症状,多无明显症状[24]。因此PCD被认为是预后最好的一种疾病,值得早期筛查。本次诊断SCADD、MCADD、VLCADD、CPTⅠ和CPTⅡ各1例。SCADD是因为短链酰基辅酶A脱氢酶基因缺陷,造成丁酰肉碱和尿中乙基丙二酸的异常蓄积,多起病于5岁以内,发病过程不可预测,以发育迟缓最为常见。MCADD 是由于中链酰基辅酶A 脱氢酶功能缺陷导致中链脂肪酸β氧化受阻,进而引起能量生成减少和毒性代谢中间产物蓄积,早期嗜睡呕吐,常迅速发展为昏迷或死亡。VLCADD是由于极长链酰基辅酶A 脱氢酶基因先天性缺陷所致,常在新生儿和婴儿早期发病,表现为心肌受累、死亡率高。以上5种疾病我国已有相关报道[25-27],发病率低于PCD,这些疾病在海南省的发现,丰富了新生儿筛查遗传代谢病的数据库,提高了新生儿人群筛查遗传代谢病的价值。
本研究发现的SCADD 和VLCADD 病例的基因变异均为罕见型变异。SCADD中c.889C>T突变(编码区第889号核苷酸由胞嘧啶变异为胸腺嘧啶),导致氨基酸改变p.R 297 C(第297 号氨基酸由精氨酸变异为半胱氨酸),为错义突变。PM2+PP2+PP3分析显示在正常人群数据库中的频率为0.00001,为低频变异;该基因很少有良性错义突变,错义突变是疾病的普遍机制;生物信息学蛋白功能预测软件 SIFT、PolyPhen_2、MutationTaster、GERP++、REVEL均预测为有害;HGMD数据库未有该位点的相关性报道,Clinvar数据库无该位点致病性分析结果。根据ACMG 指南,该变异初步判定为临床意义未明。VLCADD患儿的两个突变位点在正常人群中突变频率均为“–”,c.664G>A(编码区第664号核苷酸由鸟嘌呤变异为腺嘌呤)导致氨基酸改变p.G222R(第222 号氨基酸由甘氨酸变异为精氨酸),为错义突变。该突变不属于多态性位点,在人群中发生频率极低。HGMD专业版数据库已报道与VLCADD相关。经家系验证分析,受检人的父亲该位点为杂合变异,受检人的母亲该位点无突变。c.1819T>G(编码区第1819 号核苷酸由胸腺嘧啶变异为鸟嘌呤),导致氨基酸改变p.C 607 G(第607 号氨基酸由半胱氨酸变异为甘氨酸),为错义突变。该突变不属于多态性位点,在人群中发生频率极低。在HGMD 专业版数据库中未见报道。经家系验证分析,受检人父亲该位点无突变,受检人母亲该位点为杂合突变。
本研究尚存在一些不足,因新生儿遗传代谢病串联质谱筛查目前在海南省是收费项目,以及部分家长缺乏对遗传代谢病及其筛查意义的了解,造成该项目的覆盖面较低,筛查阳性者的召回率达不到100%。后续我们将继续加强遗传代谢病的宣传教育,提高串联质谱筛查的覆盖面和召回率。
综上,本研究结果表明FAOD 在海南省少数民族地区新生儿人群中的发病率并不低,其中PCD是该地区常见的FAOD 疾病,本研究可为相关政府部门在海南省新生儿人群中实施多种遗传代谢病筛查的决策提供基础数据。
致谢:感谢中国出生缺陷干预救助基金会对海南省新生儿多种遗传代谢病检测项目的资助。
