腺苷脱氨酶2 缺乏症临床特征与基因型分析
2024-02-25武亚丽
周 洋 武亚丽 丁 艳
华中科技大学附属武汉儿童医院风湿免疫科(湖北武汉 430016)
2型腺苷脱氨酶缺乏症(deficiency of adenosine deaminase 2,DADA 2,OMIM 编号#615688)是一种常染色体隐性遗传病,由腺苷脱氨酶2(adenosine deaminase 2,ADA2)基因功能丧失(loss of function,LOF)变异引起,是第一个明确的单基因血管炎综合征[1-2]。该遗传病于2014年被首次报道,其主要临床表型包括血管炎、自身炎症、免疫缺陷和血液学改变等症状。随着对该疾病的深入认识,越来越多的表型及基因型被描述[1-4]。Antoine等[5]的研究发现,被报道最多的为白种人,其次为土耳其人、格鲁吉亚人等,但是中国病例极少,这可能与国内对该病特点认识尚不全面有关。本研究通过介绍3 例ADA 2基因变异导致的DADA 2 病例,对临床特点及基因型进行归纳总结,以提高临床医师对该病的诊治能力。
1 临床资料
以2015 年1 月1 日至2019 年12 月31 日于武汉儿童医院风湿免疫科收治的 3 例DADA2患儿为研究对象。
例1,女,8岁4个月。患儿2岁时因反复发热及抽搐于外院住院诊疗,出现网状青斑样皮疹(图1A)伴随炎症指标高,予抗感染治疗后好转出院。后多次因反复发热住院,伴下肢皮疹,发热或周围环境气温升高时皮疹加重,最长间隔1~2 个月无发热,每次发热均伴有剧烈头痛及炎症指标升高。3岁时出现频繁喷射状呕吐,伴头晕、视物模糊,影像学提示脑梗死(图1 B),考虑“双眼麻痹性外斜视,陈旧性脑梗”,予以糖皮质激素后病情好转。患儿5 岁时出现左侧上肢抓持乏力,下肢行走跛行,完善全外显子检测和线粒体基因检测未检出致病性变异,予以左卡尼汀对症治疗病情好转。患儿8岁再次因“间断发热,左下肢乏力”于武汉儿童医院风湿免疫科就诊,伴随炎症指标高,脑血管CT血管造影术示动脉稍窄,予阿莫西林克拉维酸钾抗感染,地塞米松抗炎,口服氟桂利嗪诊断性治疗后好转。考虑原发病为免疫缺陷病可能,以血管炎为首发表现,再次送检基因检测示CECR1基因复合杂合突变(图2),并完善血清酶学(图3),DADA 2诊断明确。后长期予阿达木单抗联合环孢素控制原发病,长期康复训练。患儿目前病情稳定,皮疹较前明显好转。
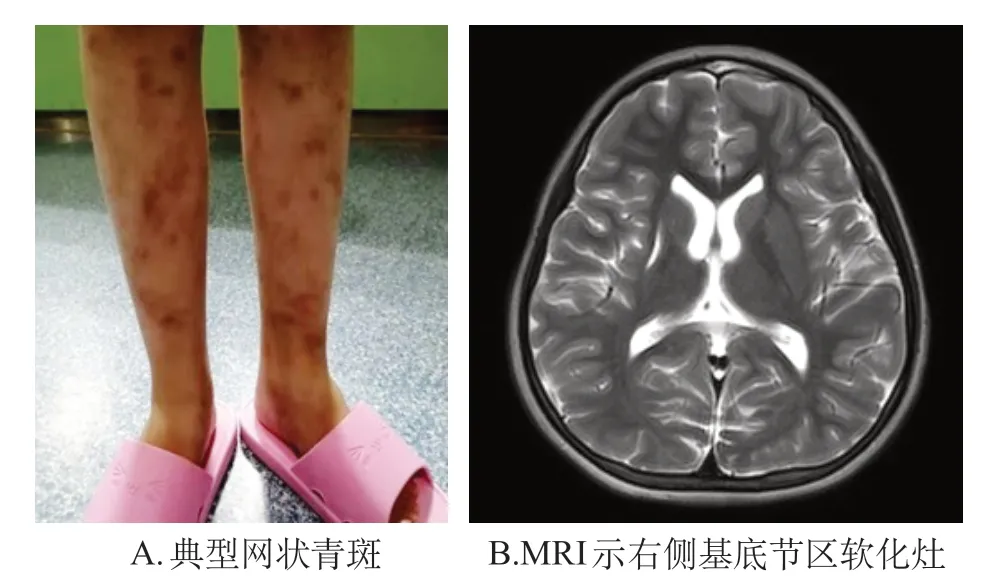
图1 例1 患儿体征和MRI 表现图
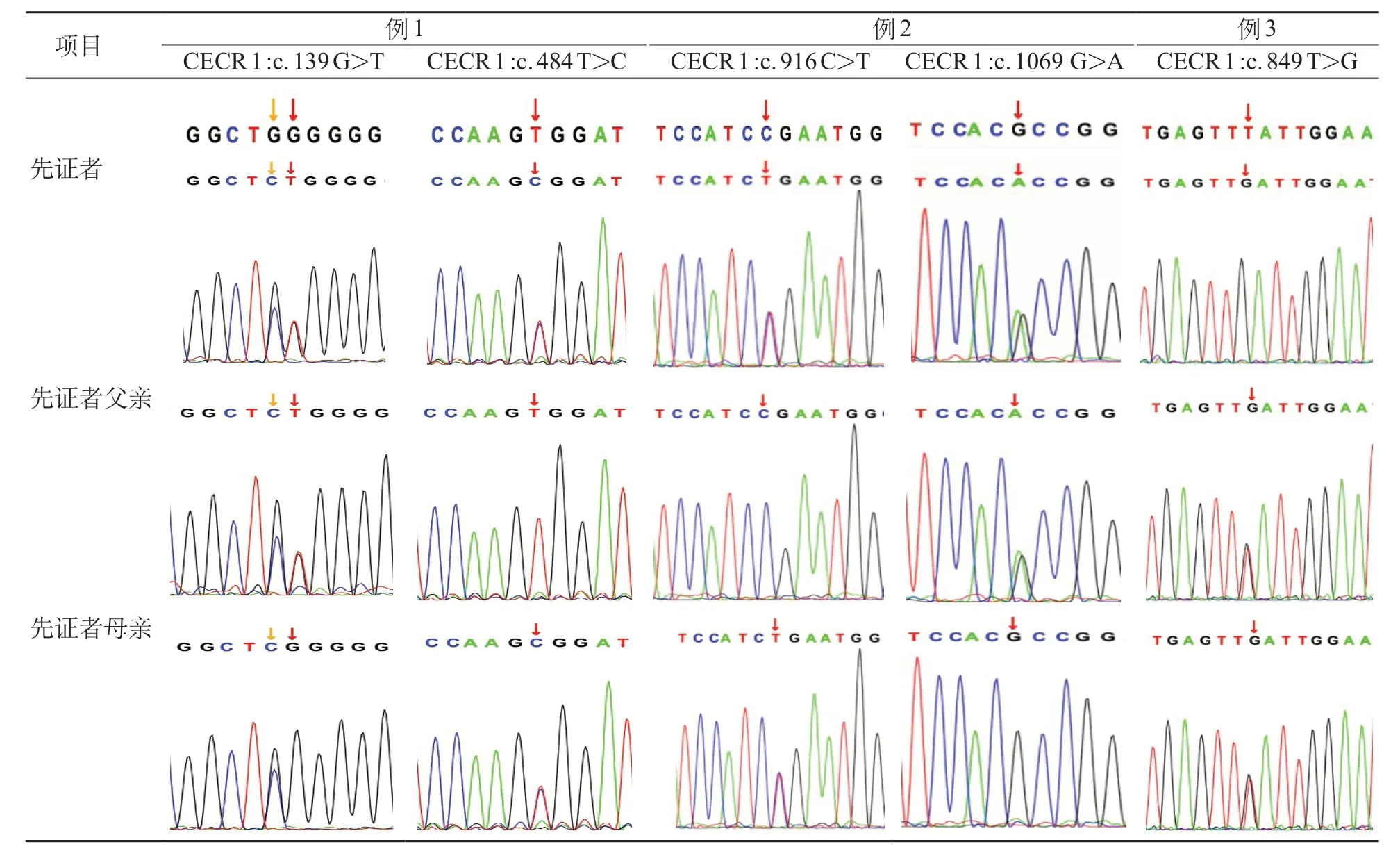
图2 患儿及其父母ADA2 基因变异测序图
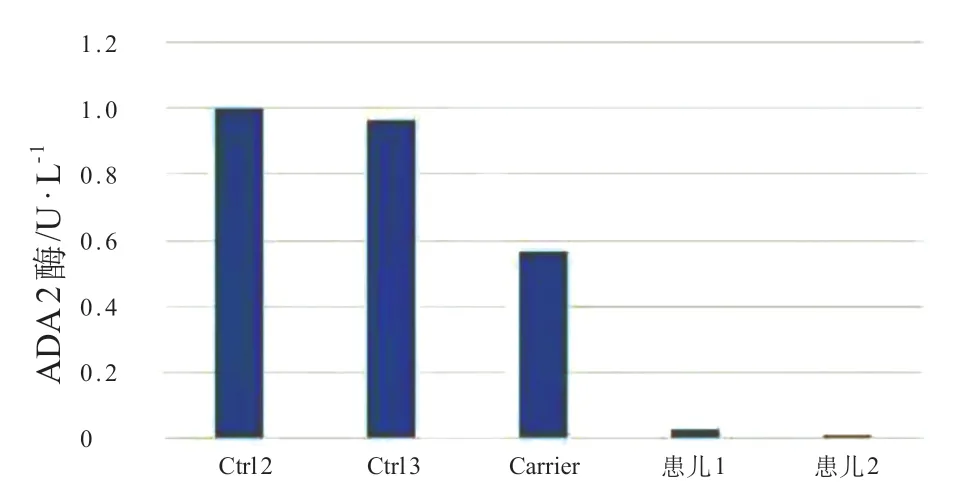
图3 血清酶学检测结果
例2,男,3 月龄,因“间断发热26 天”入住武汉儿童医院风湿免疫科,病程中全身散在红色斑丘疹,热出疹出,热退疹退,伴肝脾淋巴结肿大、炎症指标高,考虑幼年特发性关节炎(全身型),给予甲基泼尼松龙,甲氨蝶呤(5 mg/d),抗生素抗感染治疗后体温正常,仍有皮疹,出院后继续口服醋酸泼尼松,甲氨蝶呤控制原发病,但仍有反复发热、皮疹及炎症指标升高,多次调整治疗药物(甲氨蝶呤→环孢素→他克莫司,加用托珠单抗)。患儿5月龄胃肠道穿孔(图4)。腹腔探查术未见明显病灶,好转后出院。出院后继续口服他克莫司并定时返院行托珠单抗控制原发病。患儿1岁时再次因“发热1周”入住本院,病程中出现关节肿胀,辅助检查提示脑梗死(图4),完善基因检测发现CECR1基因复合杂合突变(图2),完善血清酶学(图3)。考虑诊断:DADA 2。遂予以阿达木单抗联合他克莫司控制原发病。患儿现病情稳定,定期返院复查。
例3,男,从出生开始反复发热咳嗽,平均每2个月1次,5岁时于外院行布鲁顿酪氨酸激酶(BTK)基因检查提示单核苷酸多态性(SNP),后于外院诊断“免疫缺陷病”。患儿12岁因“全身疼痛伴间断发热1月余”入住武汉儿童医院风湿免疫科,辅助检查:免疫全套(5项) IgG 2.46 g/L↓,IgA<0.24 g/L↓,IgM<0.18 g/L ↓,C 3 1.56 g/L ↑。免疫功能监测及Treg细胞计数8项CD19+%9.42%↓,CD19+130个/μL ↓,CD 3+%83.79%↑CD 3+CD 4+T%43.36%↑。结合患儿病情及检查结果,考虑为免疫缺陷病。行全外显子基因测序,报告提示CECR1相关变异(图2),结合临床表现,诊断DADA2。患儿后于外院就诊,因感染合并巨噬细胞活化综合征去世。
例1 患儿CECR 1基因外显子区域发现一对复合杂合突变,其中c.139G>T遗传自父亲,c.484T>C遗传自母亲。两个突变位点在所有正常人群数据库频率<0.0005,且两种统计方法预测变异对基因产物有影响。c.484T>C在HGMDpro和ClinVar数据库中均未见报道。c.139G>T突变已被报道。
例2 患儿CECR 1基因外显子区域发现一对复合杂合突变,突变c.1069 G>A 遗传自父亲,突变c.916C>T遗传自母亲。根据ACMG指南c.1069 G>A 突变评级为“意义不明确”,但该突变点人群数据库频率<0.0005,反式位置检测到致病变异,且两种统计方法预测出突变对基因产物有影响。c.916C>T突变等级为“致病”;在HGMDpro和ClinVar数据库中已被报道。
例3 患儿在22 号染色体CECR 1基因外显子区域发现一对纯合突变,患者检测到CECR 1基因c.849T>G纯合突变,父母均为杂合,该错义突变位于致病热点区,且正常人群数据库频率<0.0005,突变为不确定。该位点未被报道。
2 讨论
DADA2的致病基因位于染色体22q11.1,可编码ADA2蛋白[2]。该蛋白是1978年被分离发现的二聚体分泌蛋白,在人体中具有腺苷脱氨酶活性,可催化腺苷和2’-脱氧腺苷分别转化为肌苷和2’-脱氧核苷[6]。ADA2主要表达在活化的单核细胞、巨噬细胞和树突状细胞上,因其受体不明确,故DADA2的致病机制尚不清楚,目前推测可能与生长因子功能、催化功能以及免疫功能障碍有关[6]。自2014年首次报道该病至今已有300余例DADA2患者被报道,且其临床表型及基因型在不断丰富中[7]。
DADA2的临床表型主要包括:血管病和炎症、免疫缺陷和血液系统异常[8-10],还有部分病例为轻症及无症状者,在家族性筛查中被发现和诊断[11-13]。血管炎及血管病主要表现为:反复发热、皮疹、网状青斑,多器官炎症损伤,炎症指标明显升高(CRP、ESR、WBC等)[9]。脏器受累中尤以神经系统表现最为常见,可表现为出血性脑卒中、颅神经及周围神经病变[14-17]。免疫缺陷主要以轻度体液免疫功能缺陷为主,主要表现为记忆性B淋巴细胞减少、低IgM水平,易发生感染及疫苗反应等[18]。血液系统异常主要表现为淋巴细胞减少、贫血,偶见有骨髓纤维化、白血病报道[19-20]。本研究总结了国内的DADA2患者,其中10 例患者(100%)均有反复发热,炎症指标升高,9例(90%)有皮肤改变,9例(90%)有神经系统改变,7例(70%)有关节痛,9例(90%)有免疫功能低下,血液系统受累7例(70%),发育迟缓2例(20%),消化道受累1 例(10%)。神经系统受累发生率大致与国外文献报道相同。其他系统发生率均高于国外,这可能与国内报道病例较少及种族特异性有关。由于DADA2好发于儿童,有血管炎表现,易出现免疫缺陷,常被误诊为幼年特发性关节炎(全身型),结节性动脉炎或免疫缺陷病,例1、例3 长期被认为是免疫缺陷,例2被诊断幼年特发性关节炎(全身型),因此在出现不明原因关节炎合并免疫缺陷及皮疹和神经系统症状时应考虑DADA2,并积极行全外显子基因测序。
DADA 2 的基因型自2014 年被发现至今,已经鉴定出至少140 个ADA 2基因变异,ADA 2基因变异类型绝大多数为错义突变(83.4%)[1],其他移码突变、剪接缺陷和基因缺失也有报道[21-23]。常见的ADA2致病变异体为p.G47R、p.G47A、p.R169Q、p.Y 453 C 和p.T 360 A,其中p.G 47 R 和p.R 169 Q 纯合突变最常见[24]。Pui等[19]的研究发现,DADA2患者基因型和表型之间可能存在一定相关性,即纯合突变临床表型异质性较强,可以从严重表型到无症状不等,意味着遗传因素、修饰因子、环境等共同决定该基因表达。本次研究报道的3 例患儿加上报道的7 例总共10 例,70%为复合杂合突变,30%为纯合突变,共15个突变位点,其中错义突变位点12个,基因缺失突变位点2个,移码突变位点1个,与国外文献报道中错义突变占比大致相同。其中本研究报道的2个位点[c.484(exon2)T>C,c.849(exon4)T>G]既往研究中未见报道,为ADA2基因新发突变位点,丰富了DADA2的基因型。
DADA2的正式诊断标准尚未建立。临床、实验室和影像学发现以血管炎、免疫功能失调和血液系统异常为特征的自身炎症性疾病的患者,应怀疑存在DADA 2 并进行ADA 2基因测序和ADA 2 血浆蛋白水平或活性检测。如发现基因功能丧失的ADA 2致病性变异体和较低的ADA 2 蛋白活性(低于正常水平5%),可以确立DADA2诊断[19]。
该病治疗因疾病的主要表现和严重程度而异。对于有症状的患者,目前有类固醇、环磷酰胺、硫唑嘌呤、静脉注射免疫球蛋白(IVIG)和抗细胞因子治疗。其中类固醇被广泛使用,但大多数患者对高剂量类固醇有部分反应,减量后复发,一些患者可能对类固醇无效[1]。目前首选TNF-α拮抗剂,其在控制全身炎症和血管炎表现方面非常有效,但在血液学异常方面无效[25]。阿达木单抗和依那西普是最常用的[26],一些患者可能需要比平常更高的剂量才能充分控制疾病。对于无反应的患者,可考虑从单克隆抗体(阿达木单抗、英夫利昔单抗和戈利木单抗)和融合蛋白(依那西普)切换。其他细胞因子抑制剂,如IL-1 阻滞剂和IL-6 阻滞剂已经在少数患者中尝试过,大多数研究报道对IL-1受体阻滞剂无反应[27],虽然托珠单抗可有效控制炎症,但易出现病情复发[12-13]。对于难治性血液学异常DADA 2 患者,造血干细胞移植是潜在治愈性治疗策略[28]。本研究3例患儿,其中2例及时诊断后予以糖皮质激素及免疫抑制剂后症状无明显改善,予以阿达木单抗后病情平稳,可证实TNF-α拮抗剂的有效性。
综上所述,此报道总结了国内DADA 2 患儿的临床及基因型特征,该病临床表现异质性强,基因多变,对起病早,反复发热、皮疹、网状青斑、有结节性动脉炎样改变,合并脑卒中、免疫缺陷或血液系统损害的患儿,应当考虑此病,并及时完善血清酶学测试及基因检测明确诊断。本研究中,DADA2于婴儿期出现消化道穿孔表型为国内首次报道,2个位点为国内外首次报道的ADA 2基因新位点,丰富了该疾病的表现形式。
