STXBP1 基因相关脑病患儿的临床表型和基因变异分析
2024-02-25李小丽张晓莉贾天明
李小丽 张晓莉 李 肖 韩 瑞 徐 丹 甘 玲 贾天明
郑州大学第三附属医院小儿神经内科(河南郑州 450052)
发育性癫痫性脑病(developmental and epileptic encephalopathy,DEE)是一组神经发育性疾病,以早发癫痫、脑电图异常、发育迟缓或倒退为主要特征,DEE病因复杂,遗传性病因占到50%以上[1-2]。
突触融合蛋白结合蛋白-l (syntaxin-binding protein 1,STXBP 1)基因位于9 q 34.11,编码STXBP1蛋白,该蛋白对整合膜蛋白受体复合物的形成及突触囊泡融合过程至关重要[3]。因此在神经元突触信号传递过程中起着重要的调节作用。2008年Saitsu 等[4]首次在5 例大田原综合征患儿中发现致病STXBP 1基因变异,随后多项研究证明,STXBP 1基因变异与West 综合征、Dravet 综合征、未分类的早发性EE、非综合征性癫痫、孤独症谱系疾病、Rett综合征及智力残疾有关,认为STXBP 1基因变异相关疾病是复杂性神经发育疾病,统称为STXBP 1脑病[5-9],目前国内报道病例数尚不多,本研究回顾性分析2015 年10 月至2022 年5 月郑州大学第三附属医院收治的11 例STXBP 1脑病患儿,对所收集的临床资料及基因结果进行分析,并进行随访,以提高临床对该类疾病的认识,指导临床早期诊断及治疗。
1 对象与方法
1.1 研究对象
选择2015 年10 月至2022 年5 月郑州大学第三附属医院收治的11例STXBP1脑病患儿资料进行回顾性分析。研究方案获得郑州大学第三附属医院医学伦理委员会批准,伦理批号:(2020)医伦审第57号;患儿监护人均签署知情同意书。
本研究纳入标准,满足以下5 条。①年龄≤14周岁;②临床表型满足其中之一:频繁的癫痫发作、脑电图示发作间期频繁的癫痫样放电、精神运动发育落后或发育倒退;③病史资料完整,已完善头颅磁共振成像(MRI)、脑电图(EEG)及其他相关检验检查;④患儿基因检测结果根据ACMG指南提示存在STXBP1基因疑似致病或致病变异;⑤治疗规范、依从性好。
排除标准:①围产期脑损伤;②中枢神经系统感染、缺氧缺血、脑外伤、脑肿瘤;③血串联质谱和/或尿气相色谱-质谱检查提示代谢病;④家族病史与既往病史记录不完整。
1.2 研究方法
收集并分析11例STXBP1脑病患儿的临床资料:包括性别、年龄、癫痫首发年龄、发作类型、治疗过程、个人史、家族史、发育情况、脑电图、头颅MRI及基因结果等,并通过门诊复诊及电话随访收集患儿治疗及预后情况。
随访资料包括,①癫痫发作情况。②治疗情况:抗癫痫药物及其他治疗手段 (生酮饮食、迷走神经刺激术及手术治疗等)。③发育情况:根据家长提供的信息对患儿进行智力和运动的评估。除通过查询门诊信息系统获取随访信息外,必要时电话联系患儿家长。
癫痫发作患儿所使用的抗癫痫药物疗效判断标准[10]:疗效包括完全缓解(无发作)、显著有效(发作减少>75%)、部分有效(发作减少超过50%~75%)、无效(发作减少<50%或者发作增多),其中前三者统称为有效。初始疗效判断时间为药物剂量达目标剂量后观察发作间期时长的3 倍时长,发作频繁甚至每天发作者,治疗达目标剂量2 周后即可判断疗效。
药物难治性癫痫定义:参照2010年ILAE标准[11],即根据癫痫的发作类型,经过合理的选择并争取使用至少2 种抗癫痫药物,单药前后分别使用或者联合应用,无发作的时间未达到治疗前最长发作间隔的3倍或1年。
随访时对患儿采用Griffth发育评估量表进行评估:Z值≥-1 发育指标正常,-2 ≤Z值<-1 轻度发育迟缓,Z值<-2重度发育迟缓。
2 结果
2.1 一般情况
STXBP1脑病患儿共11例,其中男4例(36.36%),女7例(63.64%),其中10例患儿存在癫痫发作伴发育迟缓,1 例患儿仅表现为发育迟缓;2 例患儿有癫痫家族史,其中1例合并智力低下家族史;1例患儿合并重度听力损伤及视觉通路异常,1例合并肌张力障碍(表1)。
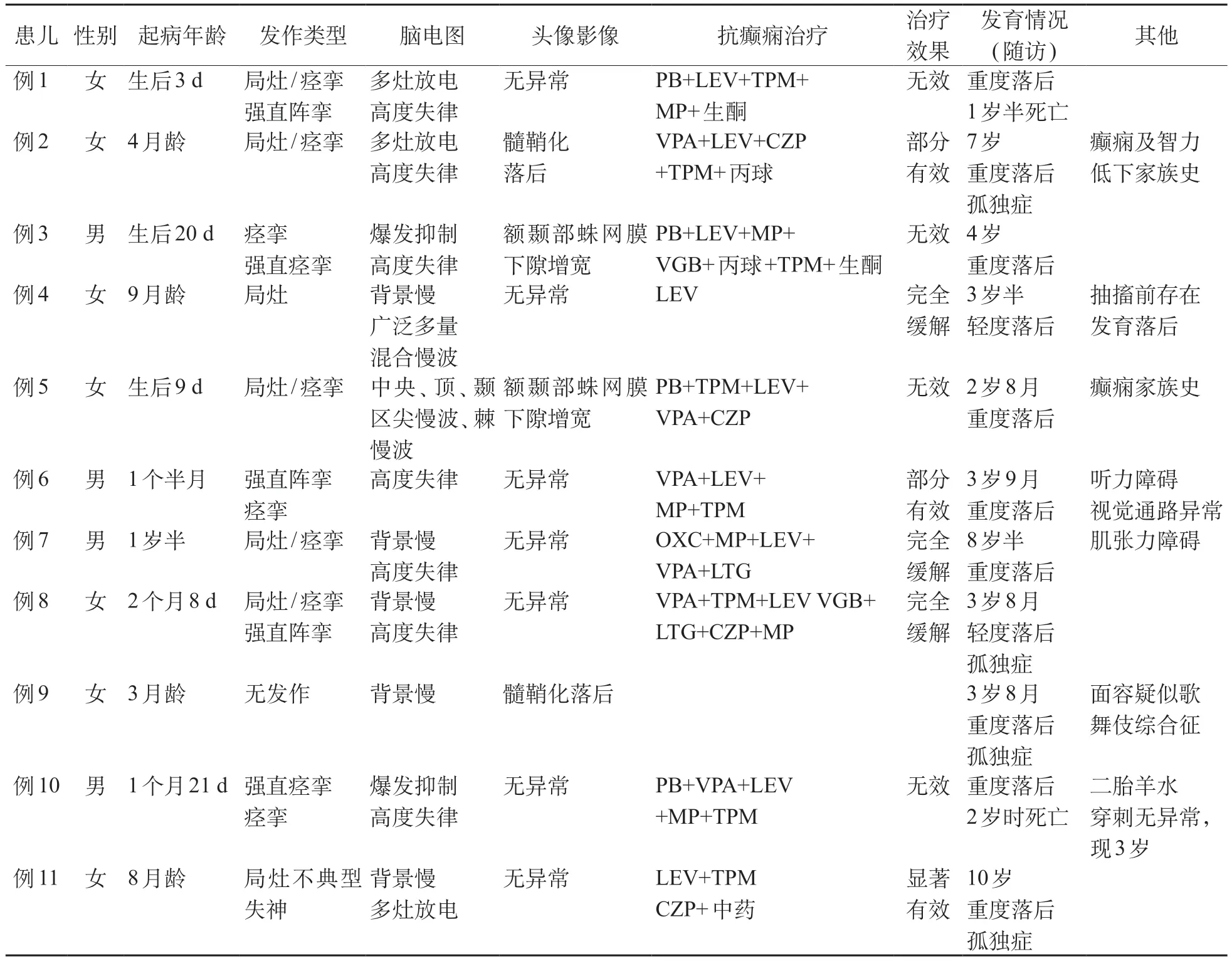
表1 11例STXBP1脑病患儿的临床资料
2.2 癫痫发作情况
首发年龄最小者为生后3天,最大者为18个月,3 个月以内起病者6 例(60%),3~6 个月起病者1 例(10%),6~12个月起病者2例(20%),12个月以上起病者1例(10%),中位年龄为59.5天,高峰年龄为l2个月以内,共9例(90%)。
发作类型及癫痫综合征:10例患儿中仅1例有1种发作类型:局灶性发作;余9例均存在2种及以上的发作类型,包括痉挛、强直阵挛、局灶性发作、强直痉挛、不典型失神,常见的发作类型为:局灶、痉挛发作。10例患儿中符合癫痫综合征诊断者7例,其中大田原综合征2例,后期转型为婴儿痉挛症,婴儿痉挛症5例,余3例不满足特定癫痫综合征诊断。
视频脑电图检查:11例患儿均完成了视频脑电图检查,9 例患儿存在发作间期癫痫样波:4 例为多灶放电,2例为爆发抑制图形,7例为高度失律图形。头颅MRI:4例患儿存在非特异性异常:2例为髓鞘化发育落后、2例为额颞部蛛网膜下隙增宽,余患儿头颅MRI均无异常。
随访时间为3 个月至6 年10 个月,随访时患儿年龄为2 岁8 个月至10 岁,2 例患儿已死亡(例1 为1 岁半时、例10 为2 岁时死亡),余均存在中重度发育迟缓,认知和语言落后显著,4例存在孤独症样表现。10 例癫痫患儿中,仅1 例应用1 种抗癫痫药物,余9例均应用3种及以上抗癫痫药物联合应用,包括PB、LEV、VPA、TPM、CZP、甲基泼尼松龙、VGB及LTG。10 例患儿均应用LEV 治疗:其中1 例为单独应用LEV 癫痫发作完全缓解,5例部分有效,4例无效。2 例患儿联合生酮饮食治疗部分有效。3 例患儿完全缓解(1例患儿单独应用LEV控制2年半无发作,1例添加CZP后发作控制,1例最初应用甲基泼尼松龙冲击后2 年无发作,后再次发作后添加LTG 后发作控制)。
2.3 基因检测结果
11 例患儿均存在STXBP 1基因新生杂合突变,其中错义突变7例、移码突变1例、剪切突变1例、缺失突变2例,复合杂合突变1例(一个位点为插入突变,另一个位点为错义突变)。根据美国遗传学与基因组学会(The American College of Medical Genetics and Genomics,ACMG)指南,其中4 例为致病性变异,7例为疑似致病性变异。移码突变和剪切突变均为致病性变异;2 例缺失突变中,1 例为位点缺失,另外1例为6-17号外显子缺失,均为疑似致病性变异。其中有7 例患儿的突变位点尚未见文献报道,分别为c.1694 T>A、c.1115 T>G、C.133_135 del、C.1543dupG、6-17号外显子杂合缺失、C.429+1G>C、C.855C>G及c.842_843insGGACGACGGCCT GTGGATAGCACT,见表2。
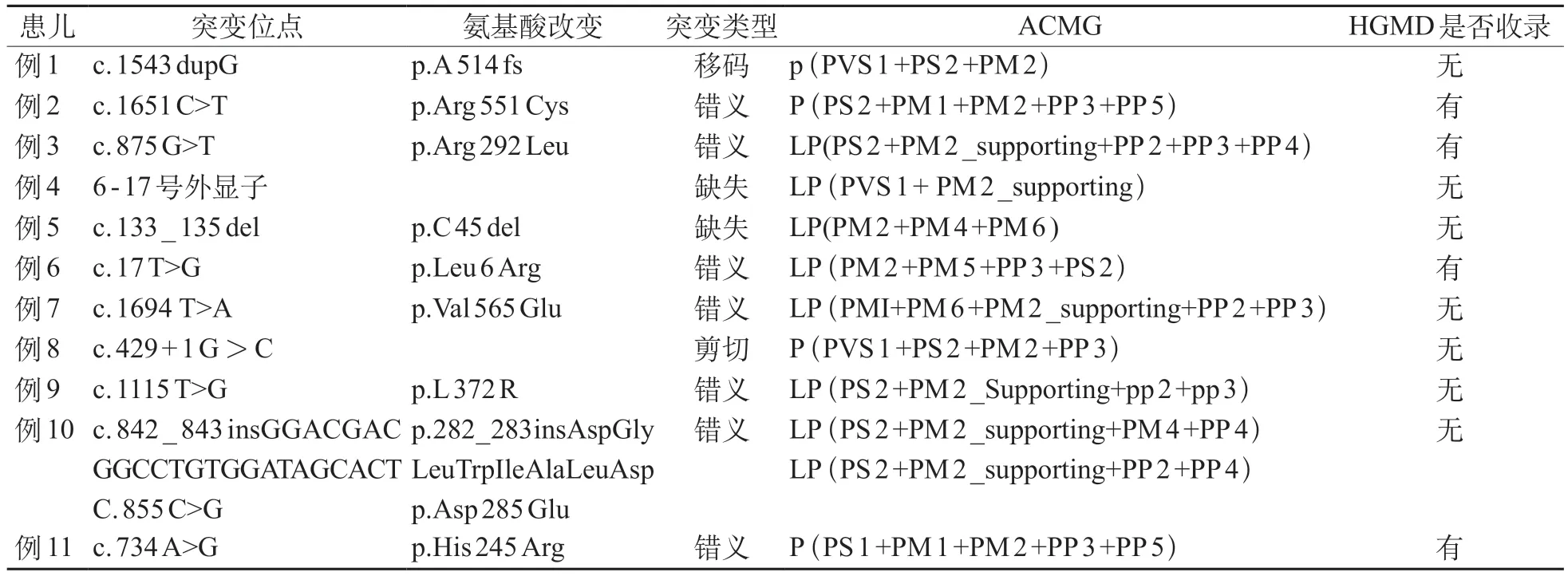
表2 11例STXBP1脑病患儿的基因变异信息
3 讨论
STXBP1基因在哺乳动物神经系统中广泛表达,编码STXBP1蛋白,参与SNARE核心复合体的形成以及突触小泡与突触前膜的融合,影响神经递质的释放[12-13]。目前STXBP 1基因变异导致STXBP 1脑病的具体机制尚不清楚,推测神经递质传输功能受损可能是其重要的损伤机制[14]。
STXBP 1功能障碍引起抑制性神经递质释放减少,导致神经网络过度兴奋,从而解释STXBP1脑病的癫痫表型。STXBP 1基因变异最初在大田原综合征中被发现,随后在West综合征[15]、Lennox-Gastaut综合征[16]及Dravet 综合征[17]中相继被发现,表明STXBP 1基因变异与癫痫性脑病关系密切。2016 年Stamberger等[18]报道147例STXBP1脑病患者,140例(95%)患有癫痫,发作类型多样,痉挛(65.3%)、局灶(57.9%)和强直发作(41.3%)占前三位,脑电图以局灶或多灶放电为主,爆发抑制和高度失律最常见;头颅MRI 近一半正常,其他患儿MRI 可表现为脑萎缩、胼胝体薄、皮质发育不良、白质髓鞘化延迟。2022年Xian等[19]统计534例STXBP1相关疾病的临床体征为:95%的神经发育异常和89%的个体癫痫发作,局灶性发作是最常见的发作类型(47%),超过88%的患者在出生后一年内发生癫痫发作。本组11例患儿中10例(90.9%)存在癫痫发作,新生儿起病占30%,3个月以内起病占60%,1岁以内起病者占90%,提示癫痫发作多于婴儿期起病;10例中9例存在多种发作类型,以痉挛、局灶及强直阵挛发作为主;脑电图异常以多灶放电、高度失律为主;11例患儿均完善了头颅核磁检查:8 例无异常,4 例存在非特异性异常;10 例患儿中7 例符合癫痫综合征诊断:大田原综合征2 例,后期均转型为婴儿痉挛症,婴儿痉挛症5例,提示婴儿痉挛症为STXBP1脑病的常见癫痫综合征表型。
STXBP1脑病的另一重要临床表型是发育迟缓,最初认为是频繁癫痫发作导致脑功能受损所致。随着研究的深入,Sivaraju等[20]发现患儿癫痫控制后,认知功能仍有明显的延迟。本组11例患儿中,9例存在中重度发育迟缓,3例患儿癫痫发作控制后运动发育有所进步,但认知和语言落后显著,仅会说2、3个字,且多为模仿性语言,主动语言及认知理解力差,与文献报道一致。Gburek-Augustat等[21]发现携带致病性STXBP1基因变异的患者可单独表现为认知障碍而无癫痫发作,表明STXBP 1基因变异所导致的癫痫与发育迟缓可能是两个独立的症状。本组11例患儿中有1例患儿仅表现为发育落后伴孤独症样表现,且容貌疑似歌舞伎综合征,其容貌特征尚未见相关文献报道,但因例数太少,不具有特异性。
除了癫痫发作和发育迟缓外,既往文献报道STXBP1基因变异患者还可出现肌张力低下、共济失调、震颤、肌张力障碍及孤独症谱系疾病,本组11例患儿中,4 例合并孤独症谱系障碍,1 例合并重度听力损伤及视觉通路异常,1例合并肌张力障碍,其具体的发病机制尚不清楚。
STXBP1基因在进化上高度保守,其发生错义、无义、剪切及移码突变均可导致功能丧失。文献报道约88%变异为点突变、缺失或插入,约12%是由于拷贝数突变,主要为缺失突变[22],包括部分或全部STXBP 1基因、甚至包含整个STXBP 1基因在内的大片段染色体缺失[19]。本组11 例患儿均存在STXBP 1基因新生杂合变异,其中错义突变7 例、移码突变1例、剪切突变1例、缺失突变2例。其中4例为致病性变异,7例为疑似致病性变异。其中有7例患儿的突变位点为首次报道,扩大了STXBP 1脑病的基因谱。Balagura等[23]总结了48例STXBP1脑病患儿的临床资料,没有观察到明确的基因型-表型相关性,认为发作年龄与发育结局的严重程度和达到的发育里程碑相关,发作时间越晚,发育结局越好。相反,发作缓解时的年龄和癫痫持续时间不影响神经发育结果,对临床医师预测患儿预后有一定提示意义。本研究2例死亡患儿均在生后3月内起病,是否提示起病年龄越早,预后相对越差,尚需大样本病例资料支持。
对STXBP1脑病的治疗策略主要是控制癫痫发作,同时兼顾发育、认知和行为等方面问题的多学科治疗[24],针对癫痫发作主要是应用抗癫痫药物,约半数患儿使用抗癫痫药后发作可完全控制,但仍有超过半数的患儿需要3 种抗癫痫药物治疗。研究报道丙戊酸、左乙拉西坦、糖皮质激素、生酮饮食[25]等可能有效,其中左乙拉西坦通过与神经突触囊泡糖蛋白2A结合,调节突触囊泡释放,抑制钙通道和细胞内钙的释放,抵消其致癫痫作用。研究报道左乙拉西坦能提高STXBP 1脑病患儿癫痫发作控制率,同时可改善患儿的脑电图[26]。本组10例合并癫痫患儿均应用左乙拉西坦治疗,其中例4 患儿单独应用左乙拉西坦发作完全缓解,脑电图正常;5例患儿部分有效,4例患儿无效。9例患儿均应用3种以上抗癫痫药物,为药物难治性癫痫,提示癫痫治疗效果存在明显异质性。
STXBP1脑病临床表型复杂,癫痫发作和发育迟缓为主要临床表型,癫痫发作起病年龄早,发作类型多样,多表现为发育性癫痫性脑病,治疗相对困难,发育迟缓相对严重,对于合并发育迟缓的癫痫患儿应选择性地进行基因检测有助于癫痫疾病的分型及个体化治疗。
