电子自旋效应在电催化剂中的作用
2024-01-22李景学于跃徐斯然闫文付木士春张佳楠
李景学,于跃,徐斯然,闫文付,木士春,张佳楠,*
1郑州大学材料科学与工程学院,郑州 450001
2郑州大学材料科学与工程学院,郑州市先进能源催化功能材料制备技术重点实验室,郑州 450012
3吉林大学化学学院,无机合成与制备化学国家重点实验室,长春 130012
4武汉理工大学,材料复合新技术国家重点实验室,武汉 430070
1 引言
有限化石能源燃烧引发的有害气体排放增多,导致环境问题日趋严重,并且随着我国“双碳”目标的提出,迫切需要开发可持续清洁能源转换技术1–5。许多能源转换技术(如光电转化6,7、电解水8、燃料电池9、金属空气电池10,11、二氧化碳和氮气还原12,13)被开发出来以缓解能源危机和环境污染。高性能电催化剂是这些能源转换技术得以实际应用的关键。其中几个关键的电化学反应的电催化剂研究备受关注14,15。
(1)氧还原反应(ORR)。清洁型能源质子交换膜燃料电池以H2/O2作为燃料,利用电化学反应产生电能,反应产物只有清洁的H2O,是一种高效且清洁的能源转换器件16,17。其阴极的氧还原反应缓慢的动力学决定了电池系统的性能,缺少高效稳定的ORR催化剂是限制燃料电池发展的瓶颈问题18。目前,此类ORR催化剂主要以过渡金属氮碳(M-N-C,M: Fe,Co,Ni,Cu)和贵金属合金催化剂为主,然而其活性和稳定性的提升仍然需要对催化剂的服役机制更清晰的了解19–22。
(2)析氧反应(OER)。通过电解水绿色生产高纯氢气可以为燃料电池提供燃料23。析氧反应是电解水的阳极半反应,同时也是金属空气电池中重要的阳极半反应,这些技术的能量效率受到OER反应较高过电势的严重阻碍24。OER相关能源转换技术的实际应用,迫切需要开发性能优异的电催化剂以降低OER过电势,加速反应动力学25。目前最具发展前景的OER催化剂主要有过渡金属氢氧化物26、钙钛矿27、尖晶石28催化剂等。
(3)氮气还原反应(NRR)。氨气作为化工、食品、电子工业的重要原料,其传统工业生产过程(H-B工艺)需要持续的高温、高压环境,因而产生大量的能耗29。电催化氮还原反应能耗低、反应过程绿色环保,是替代传统H-B工艺最具潜力的技术之一30。然而N2在催化位点上的吸附活化以及N≡N断裂存在较大困难,且竞争副反应较强,氨气产率较低31。研究人员仍在为改进电催化剂的活性与选择性不断努力。当前用于NRR电催化剂的材料主要包括过渡金属、过渡金属合金32、过渡金属氮化物(Mo2N、VN)、氧化物(MoO3、Nb2O5)33、硫化物(MoS2)34以及N或B掺杂碳材料等35。
(4)二氧化碳还原反应(CO2RR)。利用电能将二氧化碳(CO2)转化为各种增值有机化合物是减少碳排放和固碳的理想方式。电催化二氧化碳还原反应涉及多个电子和质子转移过程36,37,目前存在竞争副反应严重,法拉第效率低,产物选择性差等问题。因此,合理设计CO2RR催化剂尤为重要38,39。目前CO2RR催化剂主要包括金属单晶和多晶材料,结晶多孔材料(如金属有机框架(MOFs,metal organic frameworks)40,41和共价有机框架(COFs,covalent organic frameworks)42及其衍生碳基材料也受到广泛关注。
总之,电催化剂的开发在当前能源转换与存储技术中至关重要43。近十年来,人们逐渐理解电催化的内在反应机理、结构-性能关系,在构建高性能电催化剂方面取得了长足的进展44。Nørskov等人提出的d带中心理论,为催化剂对反应物种的吸附强度确定了理论描述符,揭示了活性位点本征活性与电子结构之间的关系。然而d带中心理论目前还需要进一步完善,有必要对电子结构进行更深层次,更精细尺度的分析45。
电子自旋是组成物质基本粒子的内禀性质,在1922年Stern-Gerlach实验中发现银原子在真空非均匀磁场中发生分裂偏转运动形成了两条轨迹46,证实了原子在磁场中取向量子化。随后狄拉克提出了电子的相对波动方程47,将电子自旋以数学的形式得以表达。在1940年Pauli证明了自旋统计定理,明确了粒子自旋量子数和粒子统计行为的关系。至此电子自旋基础理论得以完善。随着电子自旋研究的蓬勃发展,以自旋为对象的研究覆盖了物理学48,49、化学50、生命科学51等多个学科。
催化剂电子自旋的研究更深入地描述了电子结构,有希望为催化剂设计理论带来新的发展契机。最近的研究表明改变电催化剂活性中心自旋态可以调节催化位点与反应物种的吸附强度,从而提高催化剂活性52。此外,催化剂的自旋构型还能影响电化学反应中的电荷传输,加速反应动力学53。然而,迄今为止关于电催化剂自旋调控以及自旋效应对催化剂性能的影响的相关总结和综述还很有限54,55。
因此,本文首先概述了能源催化中电子自旋效应的发展历史和应用,随后总结了催化剂活性位点电子自旋的调控方式。更重要的是,我们从热力学和动力学因素两方面分析了自旋效应在电催化过程中的可能作用机理,并且综述了四种电催化反应(ORR、OER、NRR、CO2RR)中自旋调控的最新研究进展。此外,我们还总结了在电催化中电子自旋的表征技术和理论计算方法。最后,我们提出自旋效应在能源催化领域可能的发展方向,为自旋电催化的发展提供了一定的理论指导。
2 能源催化的电子自旋相关效应
除了质量和电荷外,电子还具有一个重要的内禀性质——自旋56。电子自旋涉及两个任意方向,每个自旋的大小为±ħ/2。电子在原子或分子轨道中自旋状态被称为自旋态。一定条件下原子或分子内电子自旋状态朝向某一方向整齐排列的现象称为自旋极化57。电子自旋运动会产生自旋磁矩,因此物质的磁性与自旋密切相关58。在磁场中,电子自旋平行或反平行于磁场时,电子具有不同的能量。在化学反应中,由于选择定则,当反应物与产物以不同的自旋态存在时,反应会受到阻碍,反应的活化能增加,这种反应被称为自旋禁止反应59。ORR/OER过程不仅涉及含氧物种的电荷转移,还涉及氧物种的单重态与三重态之间的自旋翻转(图1a)。要提高ORR/OER催化剂的性能,就要克服这种量子态转变带来的反应能垒增加60。铁磁性材料中通常含有Fe、Co、Ni等元素,是开壳层体系材料,有较为明显自旋轨道耦合交换相互作用,对于自旋态的转变敏感,有可能削弱自旋禁止反应的能垒61。最新研究表明催化剂活性中心自旋态的改变会对ORR/OER催化活性产生影响62。电催化反应过程中可能的选择性自旋电子转移步骤可以通过自旋极化的催化剂加速(图1b)。同时,一些课题组通过操纵自旋极化提升了电催化剂的性能63,64。此外,人们发现自旋效应在NRR、CO2RR等电催化反应中也能够发挥作用65。虽然自旋效应在不同电催化反应中的具体作用机制也有所不同,但都可以从热力学因素和动力学因素两个方面分别总结阐述。下面我们将对电催化剂的自旋调控方法进行总结,并讨论电催化自旋效应从热力学和动力学分析两个不同维度的催化机制。

图1 (a)常见氧物种的自旋态。(b) ORR中的选择性自旋电子转移60Fig.1 (a) Spin states of common oxygen species.(b) Selective Spin Electron Transfer in ORR 60.
2.1 电子自旋调控方式
随着越来越多的研究人员在电催化研究中关注到了电子自旋的独特作用。为了深入研究电催化中的自旋效应,加强对相关机制的理解,我们有必要首先对电催化剂的自旋调控方法进行初步了解。目前,已报道的自旋调控方法主要有原子掺杂66–68、晶格应变69,70和磁场协同71–73等。常见的电催化剂以过渡金属原子为活性中心,其d轨道电子占据对于催化反应的影响最为关键。
基于配体场理论,过渡金属自旋态受到配位环境的影响,可以通过调节活性中心配位环境来调控其自旋状态,并进一步提高其电催化的本征活性。Zhai等人报道了用于ORR的S原子掺杂的Fe-N-C催化剂74。Mössbauer谱表明Fe活性中心的第二配位壳层中掺入的S原子诱导了Fe的自旋态转变,Fe1-NC催化剂中同时存在高自旋(HS,high spin)Fe2+和低自旋(LS,low spin) Fe3+两种位点,其中LS Fe3+的比例在S掺杂后得到了提高。通过准原位Mössbauer谱测试发现,随着ORR过程的进行,活性中心的自旋态由初始的LS Fe3+转变为HS Fe2+,这些结果证实了(LS) Fe3+是Fe1-S1.3NC催化剂的高效活性位点。密度泛函理论(DFT,density function theory)计算揭示了低自旋Fe中心对OH*脱附过程的促进作用,从而证明活性中心自旋态的调控能够提高ORR活性(图2a)。

图2 (a) S掺杂Fe-N-C诱导Fe中心自旋极化74。(b) LaCoO3在(100)、(110)、(111)构型下电子自旋态分布76。(c)恒定磁场下极化电子的产生示意图77。(d)磁效应精准加热催化剂活性位点示意图78Fig.2 (a) S-doped Fe-N-C induced Fe central spin polarization 74.(b) Distribution of electron spin states of LaCoO3 in (100) (110) (111) configuration 76.(c) Schematic diagram of polarized electron generation under constant magnetic field 77.(d) Schematic diagram of active sites of magnetic effect precision heating catalyst 78.
除了过渡金属基催化剂的自旋调控,碳材料的自旋态也可以通过掺杂原子进行调控。Yang等人通过实验和理论计算研究了O、S、Se、Te原子掺杂碳材料的NRR催化剂活性,计算结果表明杂原子掺杂可以促使电荷积累进而促进了N2在碳原子上的吸附,自旋密度图表明在掺杂杂原子后,非磁性的碳原子上产生了明显的自旋矩,这种自旋极化效应促进了第一次质子化形成*NNH化学步骤的进行,提升了NRR活性75。
晶体场理论表明,过渡金属配合物的晶体场会影响d轨道能级分裂,从而导致不同自旋态的出现。通过晶体结构设计控制自旋态是一种有效的策略。Tong等人报道了通过控制LaCoO3外延膜的晶格取向实现了自旋态调节76。特定晶格取向的LaCoO3薄膜使CoO6八面体发生不同程度的畸变,成功地诱导了Co3+从低自旋(t62ge0g)到中自旋(t52ge1g)的自旋态跃迁(图2b)。磁性测量得到的不同晶格取向的LaCoO3薄膜中中等自旋态(IS,intermediate spin) Co3+的比例不同,其中LaCoO3(100)的IS占87%,在三种LaCoO3薄膜中最高。相应地,LaCoO3(100)在电化学测试中表现出最好的电化学活性。
此外,自旋极化与物质磁性密切相关,因此磁场作用也是自旋调控的一个重要方法。当磁场作用于铁磁材料时,磁矩将在宏观上于外部磁场方向对齐,发生自旋极化,而在反铁磁和顺磁材料中不会发生此变化。Ren等人发现利用铁磁有序催化剂(CoFe2O4)在恒定磁场下进行自旋选择可以提高OER活性(图2c)。铁磁催化剂的自旋极化发生在OER的第一个电子转移步骤,催化剂和吸附氧物种之间发生铁磁性交换相互作用,Tafel斜率从120 mV∙dec-1降低至90 mV∙dec-1。分析表明,OER过程中活性位点上的总自旋守恒,在外部磁场作用下,催化剂与反应物种间的量子自旋交换作用优化了反应动力学77。
Liu等人设计了具有热分化超晶格特征的非贵金属有机框架(MOFs),利用低导热的有机分子构建隔热层,通过磁热刺激法局部处理Co、Mn磁性金属离子。隔热层限制了磁性结构的晶格畸变,同时磁交换相互作用促使了磁性离子的自旋翻转与重构,磁热处理后活性中心Co3+由高自旋态转变为低自旋态(图2d),自旋态的变化促使了活性中心电荷密度的变化,优化了反应过程中的自由能,进而调控了OER反应活性78。
活性中心价态的调节是对电子结构进行调控的有效策略,由于价态转变带来的轨道占据改变能够精确调节自旋态。Gong等人通过改变合成过程中的气体环境来操纵共价有机骨架催化剂(COF-367-Co)中Co的自旋态79。在N2气氛下合成的催化剂活性中心为COII(S= 1/2),而在O2气氛下合成的催化剂中钴为COIII(S= 0)。COF-367-Co的电子顺磁共振(electron paramagnetic resonance,EPR)谱证实了COF-367-CoII中存在未配对电子,而COF-367-CoIII中没有未配对电子,证实了Co中心自旋态的成功改变。价态调节能实现对自旋态的精准调控,为自旋电催化效应的研究提供了有力证据。
界面效应是催化剂性能调控的有力手段。核壳结构催化剂界面处的强相互作用可以建立稳定的自旋排列,这被称为自旋钉扎效应。Xu等人通过低水平硫化设计了铁磁CoxFe3-xO4尖晶石的可控表面重建,重构后催化剂达到稳定的CoxFe3-xO4/Co(Fe)OxHy构型,羟基氧化物的引入在界面处产生了强烈的自旋钉扎,在自旋钉扎作用下,简单的磁化处理进一步增加了活性位点整齐的自旋排列,OER中氧的自旋极化高度依赖于第一次脱氢产生的氧自由基,这种自旋极化使OER决速步骤发生了转移,对于降低后续的O–O耦合步骤的反应势垒至关重要。因此,自旋极化对于OER催化剂活性的增强极为关键。催化剂界面结构的合理设计能够有效实现整齐自旋排列,有助于高性能电催化剂的设计80。
基于催化剂活性中心自旋态的影响因素,已经有多种手段被开发出来以实现催化剂的自旋调控,然而这些手段尚不够精准,因此研究人员还需要继续研究完善现有自旋调控方法,并加强对自旋调控机制的理解,以实现电催化剂自旋的精准调控。
2.2 自旋效应对电催化热力学的影响
电催化剂活性中心对反应物种的吸附强度是催化剂活性高低的决定性因素。自旋构型对反应机理的热力学影响一直被忽视,然而,电子自旋相关作用,如自旋-轨道效应,常常决定了活性中心的电子结构,进而调节反应步骤中吸附能达到合适条件(既不太高又不太低)81。如(图3c)所示,在ORR/OER中处于基态下的三重态氧分子可以与铁磁性催化剂中自旋极化的电子相互作用,降低两者库仑排斥,从而降低了催化剂与反应物之间电子转移的反应势垒82。在此情况下,反应焓可以由公式(1)表示:
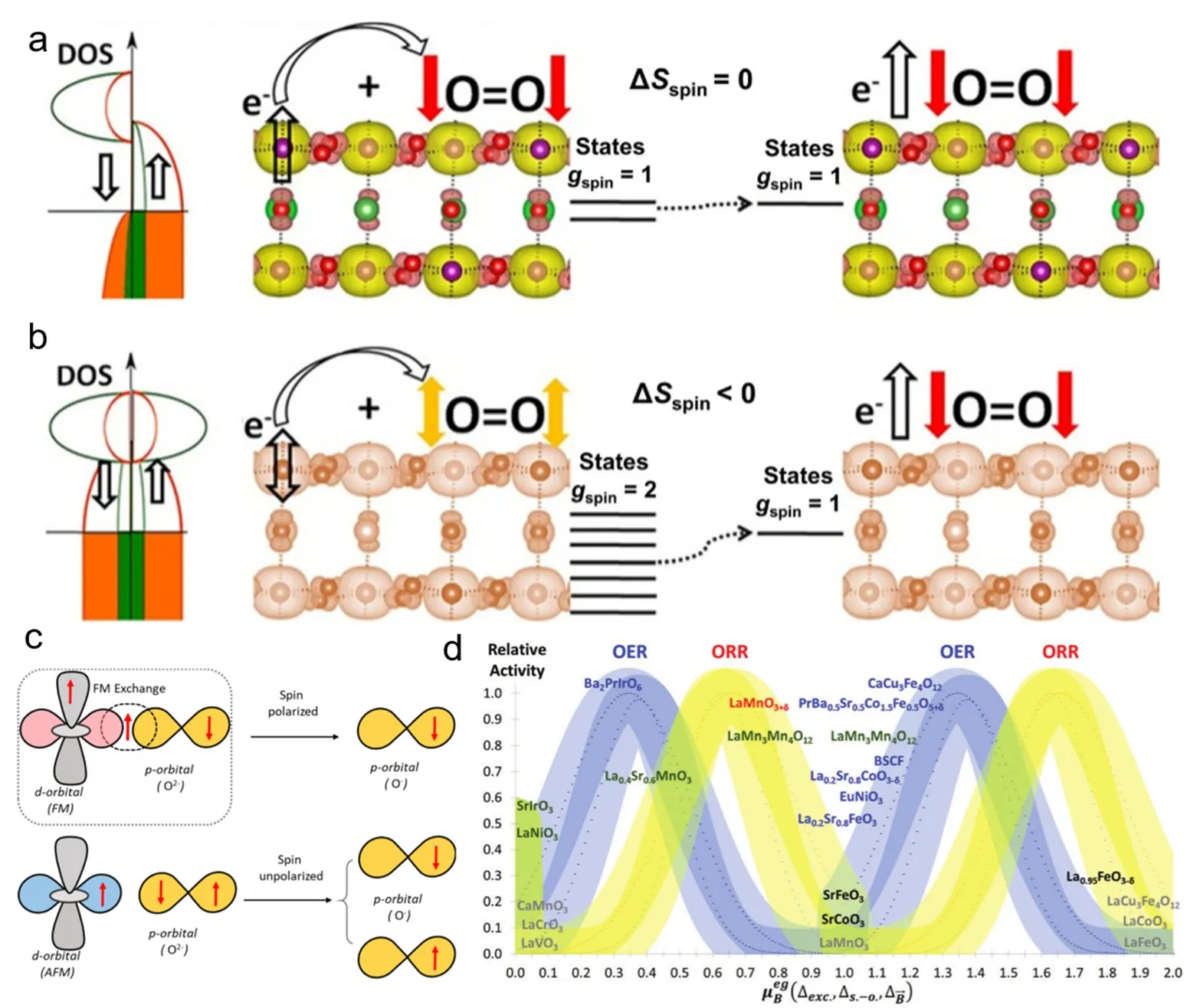
图3 (a)铁磁性氧化物和(b)顺磁性氧化物在ORR中的第一电子转移步骤的示意图85。(c) OER自旋交换机制示意图77。(d)预测钙钛矿的OER (蓝色)和ORR (黄色)活性与eg轨道磁化强度的关系86Fig.3 (a) Schematic diagram of the first electron transfer step of ferromagnetic oxide and (b) paramagnetic oxide in ORR 85.(c) Schematic diagram of OER spin exchange mechanism 77.(d) The relationship between the OER (blue) and ORR (yellow) activities of perovskite and the eg orbital magnetization was predicted 86.
其中ΔHspin是与自旋相关变化有关焓值。在反应过程中,反应机制的选择性使电子熵的增益趋于最大化,该公式表明,铁磁催化剂中有序的电子结构有助于通过交换相互作用减少与反应物的结合能,根据Sabatier原理它们的催化效率提高83。
除了反应焓(ΔH),在铁磁自旋效应还会影响催化剂的反应熵(ΔS),每个电子熵值如公式(2):式中ge-为传输电子的可能状态总数,其取决于自旋简并度()和主要来自轨道自由度的组态简并度()84。为了解释自旋对反应熵的影响,在(图3a,b)中比较了有序自旋的铁磁氧化物和无序自旋的顺磁氧化物中在ORR中第一个电子转移步骤。在该反应过程中电子由金属d轨道转移到三重态氧分子2p轨道,在铁磁氧化物中,由于稳定的铁磁电子交换相互作用而出现了自旋输运,扩展的长程磁相互作用意味着自旋简并度均为1,使得由自旋简并贡献的反应熵ΔSe-= 0。而在顺磁氧化物中,由于电子同时存在spin↑和spin↓,反应前自旋简并度为2,从而使反应过程熵减ΔSe-< 0,这使得其在热力学上是不利的。因此,电催化剂中铁磁电子有序性有利于优化反应熵,加速ORR中第一步的电子转移步骤85。
Gracia等人详细研究了钙钛矿催化剂的氧电催化活性与其活性中心磁矩的关系,得到了催化剂活性与磁矩之间的周期性火山图(图3d)。在此基础上,其进一步提出磁相互作用在最佳催化剂中将有助于电子自旋离域86。Goodenough等人发现过渡金属离子eg轨道接近1的占据态与氧键的高共价性可以提高钙钛矿型过渡金属氧化物在碱性溶液中的本征OER活性。表面过渡金属离子的eg轨道电子与表面阴离子吸附物的σ键相互作用,其eg轨道电子占据可以极大地影响氧相关中间物质在活性位点上的结合,从而影响OER活性87,88。
2.3 自旋效应对电催化动力学的影响
自旋构型除了对电催化热力学能产生影响,对反应中动力学也有重要作用。电催化过程中反应物/中间体的吸附作用和活性中心之间的电子转移以及催化剂内部的电荷传输是影响反应动力学的重要因素。电催化中电子转移会伴随着大量自旋电流的传输,因此催化剂活性中心电子构型差异所表现出电导率和电阻亦会有很大差别89,90。
磁性材料在反应过程中的电子相互作用受到量子关联,量子-电子相互作用与催化过程和效率密切相关。量子自旋(-轨道)交换相互作用(QSEI,quantum spin exchange interactions)与未成对电子的组态有关,在闭壳和开壳催化剂中都普遍存在。QESI对重塑轨道和催化结构具有重要作用,是影响键断裂和形成以及电子传输的重要因素,理解QSEI对于全面理解自旋效应在电催化中作用至关重要。(图4a)表示了量子自旋交换相互作用的时空费曼图,两个电子(φaα,φbα)分别处于两个独自轨道,在t= 0时刻两电子距离最远,随后由于电子运动开始彼此接近,在这个时候电子之间的排斥库仑势增加,随着排斥力不断增加,两电子在t1时刻交换了轨道并逐渐相互远离。在QSEI作用下,两个自旋方向相同电子通过交换轨道减少两者之间的排斥力,进而可以稳定存在。
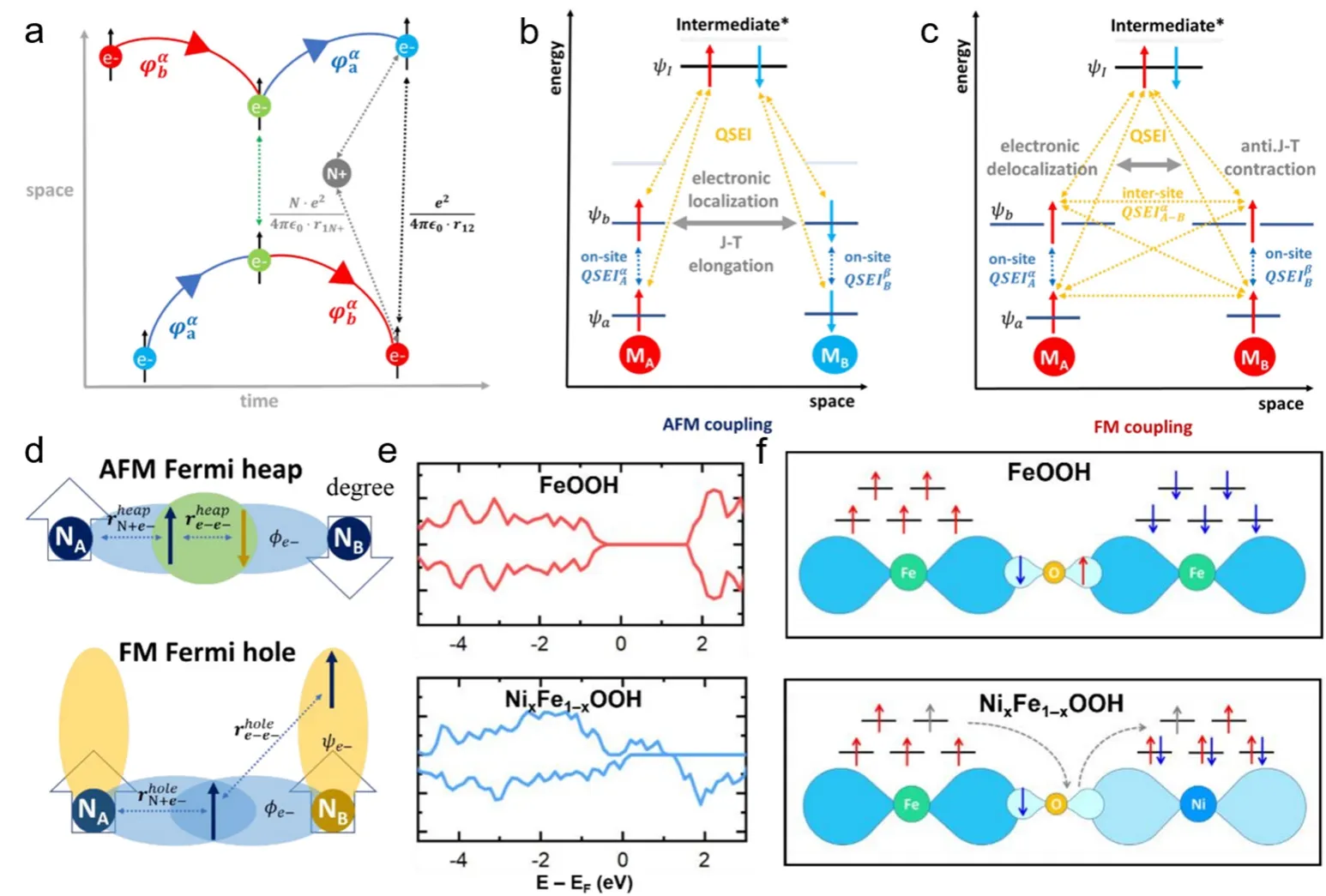
图4 (a)量子自旋交换相互作用示意图。反铁磁(b)或铁磁(c)催化剂QSEIopenshell能量/空间图。(d)反对称波函数的电子描述93。(e) FeOOH和NixFe1-xOOH的态密度图。(f) FeOOH和NixFe1-xOOH的自旋电子转移路线94Fig.4 (a) Schematic diagram of quantum spin exchange interaction.(b,c)QSEIopenshell energy/space diagram of antiferromagnetic or ferromagnetic catalyst.(d) Electronic description of antisymmetric wave function 93.(e) Density of states from FeOOH and NixFe1-xOOH.(f) Spin electron transfer route of FeOOH and NixFe1-xOOH 94.
开壳层结构(有一个以上亚层未充满的电子)催化剂在化学反应中电荷传输受到电子自旋影响。在铁磁和反铁磁结构中,反应物与中间体之间相互作用差异明显,在铁磁催化剂中由于外层电子spin↑和spin↓总数不同,原子间QSEI数量增多(图4b),铁磁活性中心价带上的开放中心产生了费米空穴,从而加快了自旋离域的交换并减少了电子排斥,如(图4c)有相同自旋的电子可以更频繁的交换轨道,这种额外的稳定能量与空价轨道一起通过量子相互作用增强催化活性。在反铁磁(AFM,anti-ferromagnetism)耦合自旋组态之间没有额外的原子间QSEI开放壳层,催化剂中仍存在原子间库仑排斥(图4d)。通常,反铁磁中常出现Jahn-Teller效应以帮助减少电子排斥91。但反铁磁材料催化反应过程中Jahn-Teller效应通常发生在高能级的反键轨道,使得最低未占轨道的不稳定化从而不利于电子传输,结果显示出反铁磁材料催化效果比铁磁材料差92。
最近,Gracia及其同事发现,在OER和ORR过程,由于未成对电子的数量不守恒,最佳磁性催化剂中的自旋势充当选择性门以影响局部自旋电流的传输。具有铁磁(FM,ferromagnetism)耦合的磁性氧化物(MO,fagnetic oxide)比具有反铁磁(AFM)自旋耦合的磁性氧化物表现出更高的电子电导率,能加快催化反应中动力学过程。OER催化活性会随着原子间铁磁耦合的增强而提高,进一步表明自旋电荷传输迁移率与催化反应活性之间不可分割的联系93。
Xu等人通过分析铁和镍阳离子的局域自旋组态,发现NixFe1-xOOH中高自旋Fe3+和低自旋Ni3+之间相互耦合产生了铁磁下的QSEI,促使NixFe1-xOOH整体呈现出高自旋态密度,并促进了催化剂内部长程电荷传输,并且沿着晶格创建了自旋通道(图4e,f),最终促使NixFe1-xOOH催化剂显示出了优异的OER活性94。
从热力学角度来讲,催化剂的活性中心自旋态能通过影响d带中心从而优化吸附能95。从动力学角度来讲,自旋电子能够降低自旋禁止反应的额外反应活化能,促进自旋电荷传输。总之,自旋效应为电催化剂的性能突破提供了新的理论指导。
3 能源催化中的自旋调控研究进展
随着自旋效应在电催化中的研究不断深入,自旋效应的热力学与动力学作用在越来越多的电催化反应中得到验证。由于反应物、产物、以及反应环境的不同,自旋效应在不同的电催化反应中的作用机理需要分别梳理。
3.1 氧还原反应中的自旋调控
Fe、CO、Ni等磁性金属中心催化剂是被研究最多的贵金属催化剂替代品,也常被用于降低贵金属催化剂中的贵金属含量96,97。磁性金属对于自旋极其敏感,探讨自旋对于ORR催化剂活性的提升是一个非常具有吸引力的课题。同时,ORR催化剂的另一个急需突破的问题是催化剂稳定性。令人惊喜的是,活性位点自旋态被证实是活性位点稳定性的影响因素98。在此,我们整理了自旋效应促进ORR催化剂活性的相关工作,并首次总结了自旋态与ORR催化剂稳定性的研究进展,希望能为高效稳定的ORR催化剂设计提供理论指导。
3.1.1 电子自旋调控氧还原反应活性
氧电催化中氧分子类型对于催化反应至关重要。通常在OER/ORR反应过程中存在单重态含氧分子化合物和三重态的氧气分子,并且单重态氧分子化合物和三重态氧气分子之间转化受到电子自旋限制。三重态氧分子具有更高自旋角动量,使其能量比单重态氧低0.8 eV99,100。因此在氧电催化中可以通过调节催化剂活性中心电子自旋排布,从而调控其在反应过程中与反应中间体的电子转移过程。例如铁磁性催化剂的单取向电子通过降低库仑排斥作用,降低了催化剂与反应物之间电子转移的反应势垒。
在氧还原反应中催化剂结构的自旋构型是催化反应的关键,通过调节金属活性中心配位原子种类或杂原子之间的长程相互作用,可以有效调节金属活性中心的自旋状态和电子结构,从而优化与反应中间体的吸附强度101。原子掺杂在ORR催化中是一种高效的调控方法。Zhang课题组利用预聚和热分解的方法制备了双金属原子分散的Fe,Mn/N-C催化剂,在传统Fe/N-C体系中引入了Mn-N基团,Mn-N基团通过自旋态跃迁和电子调制作用使得Fe3+自旋态由低自旋(t52ge0g)转变为中自旋(t42ge1g) (图5a,b,c)。Fe3+中心自旋态的变化在反应中更易于穿透氧的反键π轨道,催化剂在酸性和碱性中均表现出优异的ORR性能(在0.1 mol∙L-1KOH中的半波电位为0.928 Vvs.RHE,在0.1 mol∙L-1HClO4中的半波电位为0.804 Vvs.RHE)和良好的耐久性102。He等人将铜原子嵌入到Fe-N4相邻位置,发现铁自旋状态(磁矩)受到相邻金属Cu原子操控,Cu原子的引入减少了Fe活性中心磁矩,并且随着磁矩减少,催化剂对中间体OH*吸附能减少,在酸性和碱性ORR中均表现出更好半波电位(+0.98 V,碱性介质;+0.84 V,中性介质;+0.78 V,酸性介质),在中性/固态锌-空电池中也实现了超高开路电压(2.00 V)和大的功率密度以及电流密度(130 mW∙cm-2,186 mA∙cm-2)103。
催化剂制备过程中热解温度影响了活性中心配位结构,并且在氧还原反应中吡啶氮和吡咯氮构型对催化作用机制不同136。Zhang课题组通过自组装热解方法制备了不同N配位环境的Fe-Nx-C催化剂,通过改变热解温度调控了Fe中心电子自旋态,合成的Fe-N4催化剂具有两个平行配位的pyro-N和pyri-N (图5d),并且随热解温度变化各构型分布不同,不同的配位结构显示出Fe中心不同自旋态(图5e)。结合DFT计算,表明Fe-N4-HS结构中的3d轨道电子更容易穿透氧的反键π轨道,因此Fe-N4-HS催化剂具有更好的O2吸附能力。Fe-N4-HS催化剂显示出优异的酸性/碱性ORR活性(在0.1 mol∙L-1HClO4中的半波电位为0.916 Vvs.RHE)(图5f),以及碱性条件下显著的耐久性104。
Yan等人利用静电纺丝法将金属钴纳米点掺杂到大孔碳纳米纤维中合成了碳基的磁性催化纳米笼(MCN,Magnetic catalytic nano-cage)双功能催化剂,当对其施加中等(350 mT)磁场,由于MCN对磁场产生了限制效应,从而促进了Co原子自旋态从低自旋到高自旋的跃迁(图5g),使其具有更多未成对电子。未成对电子增多有效增强了氧中间体的吸附和反应电子转移过程,与未加磁场MCN催化剂相比ORR的半波电位增加了约20 mV (图5h,i),并且在锌空电池中0.82 V的小电压差下提供2.5倍的容量,显示出较好的可充电性和更长的寿命(大于155 h)105。
3.1.2 电子自旋调控氧还原反应稳定性
虽然在酸性燃料电池中金属-氮-碳(M-N-C)材料是一种很有前途的替代铂催化氧还原反应的催化剂,但是由于其耐久性远远不如贵金属催化剂,因此研究非贵金属活性位点的降解途径以精准提升催化剂稳定性至关重要。催化剂活性中心自旋态对于催化剂反应稳定性有着良好的调控作用106,107。Li等人研究了Fe-N-C催化剂中两个不同的FeNx位点(S1和S2)在反应过程中的降解过程,通过57Fe穆斯堡尔谱表征技术发现两种双峰(图6a,b),标记为D1 (四极分裂(QS)值为0.9–1.2 mm∙s-1)和D2 (QS = 1.8–2.8 mm∙s-1),两者具有相似的异构体位移,通过对QS值进行DFT计算,确定了D1是高自旋Fe(Ⅲ)Nx位点(S1),D2是低或中自旋Fe(II)Nx位点(S2)。随后对其进行了电化学测试,在0.5 V电压下工作5 h后,活性急剧下降,然后50 h中缓慢但稳定地下降(图6c)。结果表明活性随操作时间降低,D1的信号强度持续下降,与此同时两个六联体的信号强度(超顺磁性氧化铁)持续增加,而D2基本不变,可以确定,初期5 h工作内活性下降,是因为碳表面氧化从而降低了反应周转频率和S1位点数量,在工作5 h之后,Fe位点的周转频率稳定,导致总活性与D1或D1+D2之间的线性相关。然而,活性对D1的绝对面积图在x= 0处的外推导致正的y截距,表明催化剂阴极即使在不存在S1的情况下也应具有显著的ORR活性。总之,两种位点均有较好ORR活性,但S1在质子交换燃料电池运行中不耐用,会快速转化为氧化铁,而S2位点显示出较好的耐久性,对于质子交换膜燃料电池的应用,未来研究应进一步致力于增加S2位点的位点密度和在酸性和氧化环境中稳定S1位点108。
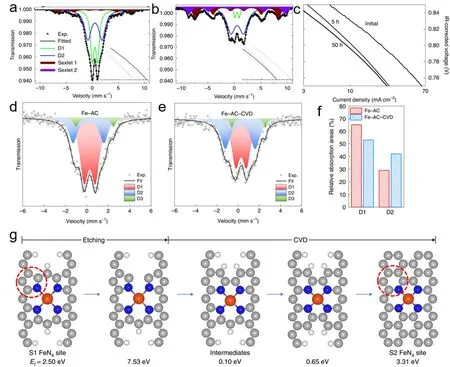
图6 Fe0.5 (a)在初始状态及(b)在0.5 V电池操作50 h后的57Fe Mössbauer光谱。(c)初始极化曲线和最终极化曲线108。(d,e) Fe-AC和Fe-AC-CVD样品的57Fe Mössbauer光谱。(f)两催化剂D1和D2位点含量图。(g)从S1到S2位点的可能结构改变途径。灰色、蓝色、白色和橙色的球分别代表C、N、H和Fe原子。Ef表示自由能106Fig.6 57Fe Mössbauer spectra of Fe0.5 (a) at the initial state and (b) after 50 h battery operation at 0.5 V.(c) Initial polarization curve and final polarization curve108.(d,e) 57Fe Mössbauer spectra of Fe-AC and Fe-AC-CVD samples.(f) D1 and D2 site content map of two catalysts.(g) Possible structural change pathway from S1 to S2.The gray,blue,white and orange spheres represent C,N,H and Fe atoms respectively.Ef stands for free energy 106.
Wu等人在高温下利用氯化铵处理Fe-N-C催化剂使其产生丰富的缺陷,随后通过化学气相沉积法(CVD)在催化剂上沉积氮掺杂的碳薄层,合成的催化剂显示更好的稳定性。通过穆斯堡尔光谱显示出Fe-N4位点局部结构,两催化剂均含有D1和D2双峰(图6d,e),对应于高活性但不稳定的S1位点(FeN4C12)和高度稳定但不太活跃的S2位点(FeN4C10)。其中沉积后的催化剂S2位点含量较高,原因可能为在高温下沉积一层薄薄的N掺杂碳导致了催化剂表面层中S1位点到S2位点的转变(图6f),随后通过DFT计算出S1位点到S2位点转变的热力学过程,计算出的S2位点的形成能比S1位点更负(图6g),表明这种转化在热力学上是有利的。据此,不难推测通过调节催化剂的局部碳结构并将不稳定的吡咯N配位S1转化为稳定的吡啶N配位S2位点,来显着提高催化剂中FeN4活性位点的固有稳定性有着一定的可行性106。
3.2 析氧反应中的自旋调控
水相中的析氧反应是提升电解水产氢效率的瓶颈。O―O键的形成涉及从反磁性氢氧化物中产生顺磁性氧分子反应过程,自旋构型可能在水相分子电催化中发挥重要作用。深入了解表面金属位点和晶格氧原子的作用对于合理设计高性能OER催化剂至关重要,其中层状双金属氢氧化物(LDH,layered double hydroxide)具有较高的自由度可以用来调节电子结构和催化性能109,110,Sun等人通过Cu2+掺杂实现了Ni-Fe-LDHs从亚铁磁体到铁磁体的转变,并精准调控了Fe3+位点自旋分裂。对于dxy平面,除了带有未成对电子的Fe3+外,Ni2+和Cu2+的dxy轨道都被填充(图7a)。因此,在掺杂Cu2+的情况下,Ni2+和Cu2+的π对称t2g轨道被完全占据,这将导致Ni2+/Cu2+与O2-之间出现电子排斥。另一方面,由于Fe3+含有未成对电子,通过Cu2+掺杂使得O―Fe―O的π轨道捐献得到加强,加速Ni2+和Cu2+的部分电子转移。即掺杂的Cu2+既可以通过电子相互作用改变电子结构,又可以促进电子转移。并且掺杂后催化剂由亚铁磁体转变到铁磁体(图7b),在磁场作用下OER性能显著提高,仅需要180 mV的过电势即可实现10 mA∙cm-2的电流密度,Tafel斜率低至33.7 mV∙dec-1(图7c)111。Kang等人利用原位电化学法将单层乙酰丙酮镍分子晶体制备了单层非晶态Ni(OH)2,结果显示单层氢氧化物比多层氢氧化物能够更容易进行脱氢反应和氧气析出反应,随后发现Co原子掺杂到体系中能够通过Jahn-Teller效应进一步调整单层LDH材料d电子结构,降低脱氢/脱氧电位并在较低电位下激活氧原子,能够进一步降低OER的过电位,确定了活性中心电子自旋排布决定了单层NiCo LDH具有优异的OER活性112。双金属基催化剂,由于其可调节的电子结构和可控的低维构造方法扩大表面积,在OER反应中有巨大潜力。Wang等人利用水热法将高价态Ta引入到了NiFe LDH,Ta的掺杂引起了NiFe LDH晶格膨胀和电子结构的改变,通过DFT计算揭示了活性中心Fe周围的电荷损失和Ta周围的电荷积累,电荷转移导致了Ta价态降低并让Ta成为OH*吸附最佳位点,优化的TaNiFe LDH表现出更好的OER活性(260 mV过电势下50 mA∙cm-2电流密度,58.95 mV∙dec-1的Tafel斜率)113。
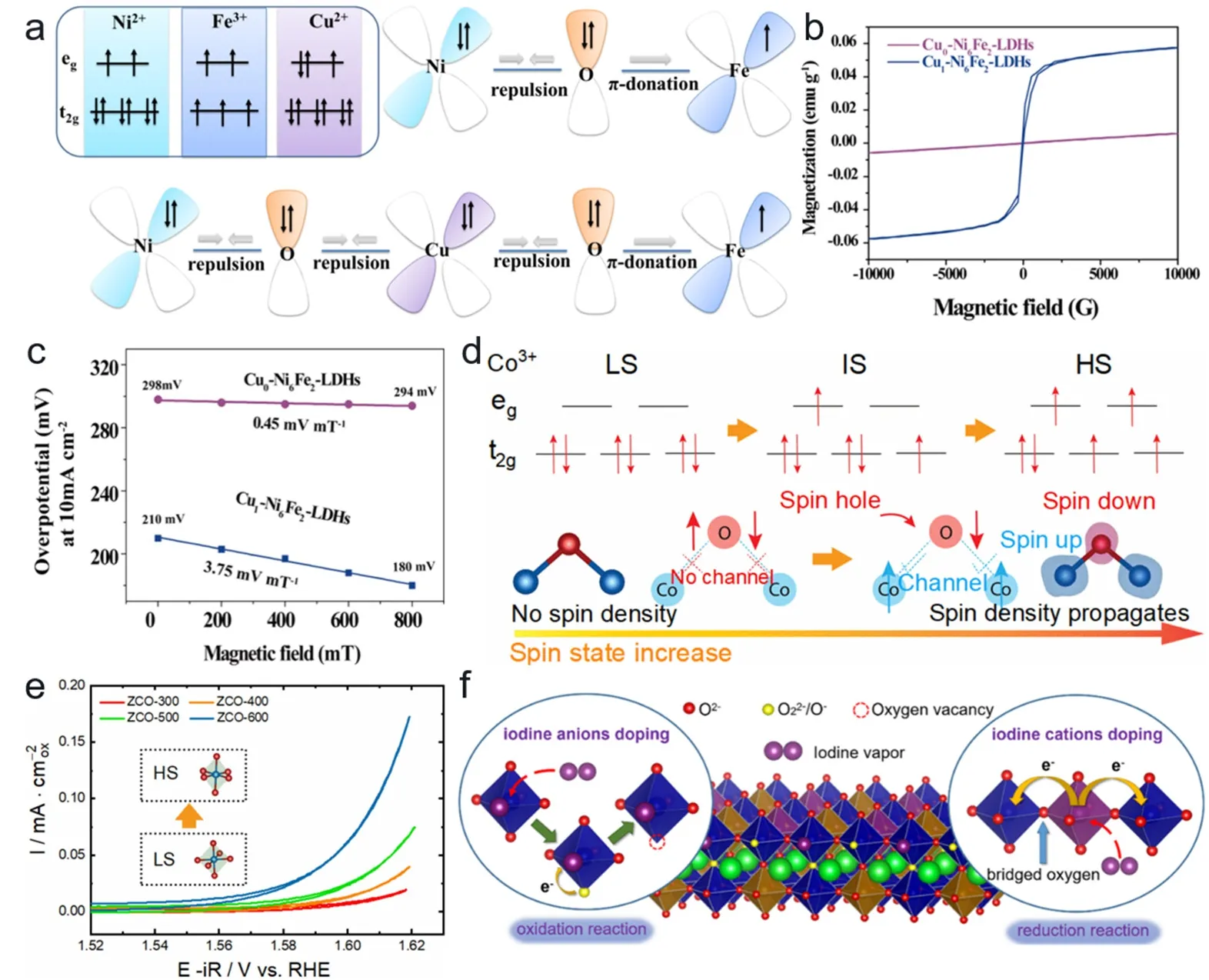
图7 (a) NiFe-LDHs和Cu-NiFe-LDH样品dxy轨道中的电子相互作用示意图。(b) Cu0-Ni6Fe2-LDHs和Cu1-Ni6Fe2-LDHs的VSM曲线。(c)磁场下Cu0-Ni6Fe2-LDHs和Cu1-Ni6Fe2-LDHs的过电位曲线111。(d) Co3+自旋态和Co-O-Co自旋通道在提升Co自旋态过程中的演化示意图。(e) ZnCo2O4样品的CV曲线115。(f) I-BSCF中碘阳离子和阴离子双位掺杂的机制示意图116Fig.7 (a) Schematic diagram of electron interaction in dxy orbit of NiFe-LDHs and Cu-NiFe-LDHs.(b) Cu0-Ni6Fe2-LDHs and Cu1-Ni6Fe2-LDHs of VSM curve.(c) Overpotential curves of Cu0-Ni6Fe2-LDHs and Cu1-Ni6Fe2-LDHs under magnetic field 111.(d) Evolution diagram of Co3+ spin state and Co-O-Co spin channel in the process of raising Co spin state.(e) The CV curve of ZnCo2O4 sample 115.(f) Schematic diagram of the mechanism of iodine cation and anion double doping in I-BSCF 116.
现有研究认为过渡金属氧化物(TMO,transition metal oxide)具有优良的OER催化活性,并且OER在过渡金属氧化物上结构-活性关系得到了理论支持114。例如,Sun等人实验中利用调控煅烧温度设计了ZnCo2O4中低自旋(LS)到高自旋(HS)态的钴阳离子(图7d),通过DFT计算确定了钴离子自旋态从低到高的转变会出现自旋通道促进电荷传输并且降低与*OOH的吸附能,实验中最优自旋态ZnCo2O4的活性优于其他典型的钴基氧化物(图7e)115。钙钛矿型(ABO3)过渡金属氧化物是很有前途的析氧反应电催化剂,但仍然存在活性不足的问题。传统的方法主要基于将杂原子掺杂到ABO3结构的B位点或O位点。Liu等人利用双位点掺杂策略以Ba0.5Sr0.5(Co0.8Fe0.2)0.9O3-δ钙钛矿(BSCF)作为模型,将碘阳离子和阴离子同时掺入ABO3钙钛矿氧化物的B位点和O位点得到碘双位点掺杂BSCF (I-BSCF)。通过表征发现,随着碘取代的增加Co原子eg轨道电子占有率达到最佳(1.2),在该自旋态下钴阳离子与氧阴离子之间共价混合,过渡金属-氧键的高共价性更有利于Co中心和吸附氧中间体之间电子转移(图7f)。导致IBSCF的OER活性增强(10 mA∙cm-2下过电势290 mV,Tafel斜率仅53 mV∙dec-1)116。
与尖晶石和钙钛矿氧化物不同的是,Zhang等人研究了新型单斜晶系ABO4氧化物在析氧反应中的催化活性,获得了单晶Fe0.4Co0.6W0.4Mo0.6O4,通过密度泛函理论计算的机理分析证实,具有低价态的八面体A位点可以作为ABO4型氧化物中的OER活性位点,并且在Co2+位点的低自旋态(LS:t62ge1g)中会发生更快的电荷转移,加速了从OH*形成O*的OER决定步骤,使其显示出优异的OER催化活性(10 mA∙cm-2下过电势276.4 mV,Tafel斜率仅30.9 mV∙dec-1)117。
3.3 氮还原反应中的自旋调控
当前氮还原反应生产氨气存在两大挑战:a)N2在催化位点上的吸附和活化以及难以打破强N≡N三键,导致NH3产率非常低;b)水性电解质中的竞争性析氢反应,导致NRR法拉第效率较低118。因此,了解活性位点的电子态与NRR性能之间的关系对于探索高效电催化剂至关重要119。催化剂活性位点的自旋态能影响中心原子电子轨道与反应物种轨道的重叠程度,通过改变电催化剂自旋态可以有效促进NRR催化活性153。
Zhang课题组将Fe和Mo两金属原子分散到聚酞菁(PPc,polyphthalocyanine)有机骨架中,形成了FeN4和MoN4配位(FeMoPPc)。研究发现与FeN4相邻的MoN4可以调节Fe中心的自旋状态由高自旋到中自旋转变(图8a)。其中Fe中心空d轨道和分离的d电子有利于Fe 3d轨道与N 2p轨道重叠,更有效地激活N≡N三键(图8b,c)。通过理论模型表明FeN4为活性位点,并且Fe自旋态的转变显着降低了决定步骤的能垒,有利于N2的首次加氢反应(图8d)。具有中自旋铁位点的催化剂(FeMoPPc)法拉第效率比高自旋铁位点催化剂(FePPc)高2.0倍120。
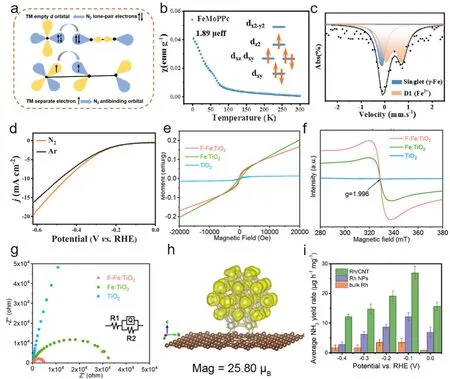
图8 (a) N2与过渡金属键合示意图。(b) FeMoPPc磁化率和电子轨道排布示意图。(c)室温下FeMoPPc的57Fe Mössbauer光谱。(d)碱性条件下,在N2和Ar气氛下LSV曲线120。(e) TiO2,Fe:TiO2和F-Fe:TiO2的M–H磁化曲线。(f) TiO2,Fe:TiO2和F-Fe:TiO2的EPR光谱。(g)样品电化学阻抗谱121。(h) Rh/石墨烯的自旋分辨密度图。黄色区域代表电荷累积。(i)不同电压下样品NH3产率122Fig.8 (a) Schematic diagram of N2 bonding with transition metal.(b) Schematic diagram of FeMoPPc magnetic susceptibility and electron orbit layout.(c) 57Fe Mössbauer spectra of FeMoPPc at room temperature.(d) LSV curves in N2 and Ar atmosphere under alkaline condition 120.(e) M–H magnetization curves of TiO2,Fe:TiO2 and F-Fe:TiO2.(f) EPR spectra of TiO2,Fe:TiO2 and F-Fe: TiO2.(g) Electrochemical impedance spectrum of the sample 121.(h) The spin resolution density diagram of Rh/graphene.The yellow area represents charge accumulation.(i) NH3 yield of samples at different voltages 122.
过渡金属氧化物,由于其催化位点配位环境易于调控而受到广泛关注,被认为是电催化氮还原反应具有潜力的催化剂材料之一。Song等人合成了表面F修饰的Fe掺杂TiO2电催化剂(FFe:TiO2),TiO2中的Fe掺杂通过局部电荷补偿诱导氧空位,导带以下的缺陷能级增强了N2吸附。表面氟改性后,Fe 3d轨道中电子向更高能级移动,电子重排后Fe转变为高自旋态(图8e,f),促使了Fe 3d电子向N2的反键合1πg*轨道转移,进而促进了N2极化和活化。此外,Fe 3d和Fe 2p的轨道杂化与电子离域,降低了N2电还原为NH3的限速步骤的能垒(图8g)。因此,F-Fe:TiO2催化剂具有最好电荷传输效率和最大氨气生产率(27.86 µg∙h-1∙mgcat.-1)121。
研究表明,具有高度自旋极化状态的催化剂可以促进N2吸附并激活其在NRR中的惰性N≡N,且贵金属的特殊电子结构对N2固定反应具有高效催化效果。Zhang等人将Rh纳米粒子锚定到碳纳米管并计算了(Rh/CNT)模型自旋态(图8h),揭示了尺寸效应相关的高自旋和与底物效应相关的电荷密度对NRR催化剂催化活性的影响。发现锚定在碳纳米管的Rh团簇会产生高度自旋极化显示出高自旋态,并且催化剂的高自旋态和Rh纳米粒子和底物之间良好电荷传输效应可以产生强烈N2吸附特性,弱化了N≡N键能,有利于活性位点和N2分子之间的电子转移。导致了锚定在CNT上的超细Rh纳米粒子表现出优异的NRR性能(图8i),尤其是高NH3产率(26.91 µg∙h-1∙mgcat.-1)和法拉第效率(23.48%)122。
3.4 二氧化碳还原反应中的自旋调控
电催化二氧化碳还原反应涉及多个电子和质子转移过程,目前存在竞争副反应严重,法拉第效率低,产物选择性差等问题123。催化剂活性位点自旋调控能够在增强CO2RR活性的同时抑制竞争副反应的发生124。最近的研究工作证明了CO2RR电催化剂自旋态与实际催化活性的相关性125。
Wang等人以氧化锌(ZnO)为模型催化剂,通过用杂原子(Mo、Cu)取代锌位点来精确调节电子状态(Mo-ZnO和Cu-ZnO)。发现杂原子掺杂导致的费米能级附近新产生的占据和未占据轨道(图9a)在同时调控CO2RR和竞争反应。Cu-ZnO中新形成的空轨道有助于增强吸附物的轨道杂化和键合,增强了*H和*HOCO的吸附(图9b),因此析氢反应活性增强,CO2RR活性下降。Mo-ZnO在价带最大处出现了两个孤立的新占据轨道,这两轨道将ZnO的占据轨道推离费米能级,并显著地下移Mo-ZnO的d带中心(-4.42 eV),削弱了吸附物的结合强度,使其拥有了最好的CO2RR活性(CO最高电流密度,-1.0 V下6.89 mA∙cm-2)。因此电催化CO2RR活性遵循Mo-ZnO > ZnO > Cu-ZnO与析氢反应活性完全相反的趋势126。

图9 (a)不同样品的态密度状态的比较图。(b)不同样品上HER和CO2 RR关键中间体结合能的比较126。(c)催化剂限制电位比较图。(d) CO2 RR吸附能与催化剂磁矩火山图130。(e) Fe-N4卟啉和Fe-N2S2卟啉上CO2还原至CH3OH和相应中间体的自由能图131。(f) Ni/Cu-N-C催化剂合成示意图。(g) Ni 3d轨道费米能级示意图132Fig.9 (a) Comparison of the density of states of different samples.(b) Comparison of the binding energies of the key intermediates of HER and CO2RR on different samples 126.(c) Comparison diagram of catalyst limiting potential.(d) CO2 RR adsorption energy and catalyst magnetic moment volcano diagram 130.(e) Free energy diagram of CO2 reduction to CH3OH and corresponding intermediates on Fe-N4 porphyrin and Fe-N2S2 porphyrin 131.(f) Schematic diagram of Ni/Cu-N-C catalyst synthesis.(g) Schematic diagram of Fermi energy level of Ni 3d orbit 132.
在已开发的单原子过渡金属锚定氮碳(M-NC)催化剂中,配位环境和过渡金属原子的电子结构是M-N-C对CO2RR的电化学活性和选择性的决定因素。调节载体的配位环境是优化单原子催化剂催化性能的有效途径127。由于高度分散的性质,单原子催化剂不仅具有最大的原子利用效率,并且其活性位点可以通过与适当的催化载体形成牢固的键合而表现出优异的稳定性128,129。Ren等人通过将Fe嵌入B掺杂的单或双真空石墨烯中,构建了一系列由B原子配位Fe单原子催化剂。合成的8种有效的CO2RR催化剂(FeC3、FeBC2、FeB2C、FeB3、FeB2C2h、FeB3C、FeB4和FeO4),它们都易激活CO2分子并显著抑制析氢反应(HER)的竞争(图9c)。研究表明,B原子的掺杂调节了中心Fe原子的d带中心和磁矩,有利于实现催化剂的活性和产物选择性。其中FeB2C拥有最负的d中心,减弱了HER副反应中间体吸附,其中Fe原子磁矩接近1.71 μB时O原子的吸附最弱,表明CO2RR催化活性最好(图9d)。其最优的磁矩和d带中心有效调控了其反应路径,产生CH4,并使其具有最低的极限电位(-0.24 V)和最佳的电催化CO2RR性能130。Cao等人以常规铁卟啉(Fe-N4卟啉)为基础,将S原子引入N配位(Fe-N2S2卟啉)对活性位点进行电子结构优化。通过密度泛函理论计算研究了两种催化剂结构稳定性和态密度,发现S原子掺杂后会在引起Fe中心费米能级附近产生新dz2轨道,有助于在Fe原子的z方向进行中间体吸附,从而提高了催化剂对CO2吸附能力(图9e)。并且S取代不仅可以增加CO的O原子中的电子密度,使CO易于氢化,还可以调节Fe的电子密度以优化CO结合,N、S配位促进吸附中间体的质子化,从而加速整体CO2RR。Fe-N2S2卟啉是一种高性能的CO2RR催化剂,其对HCOOH和CH3OH的极限电位分别为-0.38和-0.40 V,超过大多数Cu基催化剂131。
最近,有报道提出构建原子对结构是一种改变原子位点催化行为的有效方法,利用双原子位点协同电子配置效应和尺度关系提升催化活性。Zhu等人以ZIF-8为骨架制备了Ni-Cu原子对构型催化剂(Ni/Cu-N-C) (图9f)。理论模型研究表明,Cu金属掺入后,Ni 3d轨道电子移到费米能级附近,电子的跃迁促进了Ni 3d轨道和C 2p轨道之间的杂化,Ni/CuN6位上的Ni-C反键态明显上移,这种调节大大降低了速率决定步骤(RDS,rate determination steps) (*COOH形成过程)的反应势垒(图9g)。此外,Ni的不对称电子分布引起自旋极化,这也有利于中间体的吸附从而使CO2RR动力学更快。Ni/Cu-N-C催化剂表现出优异的催化活性和选择性,在-0.6 V下的转换频率(TOF,turnover frequency)达到20695 h-1,CO生产最大法拉第效率为97.7%132。
自旋效应在电催化中的作用已经得到了大量工作的证明。作为一个新兴的电催化剂研究方向,其调控方式和作用机理还需要深入研究挖掘,这必须依靠先进表征技术来实现。
4 电子自旋表征技术和模拟计算方法
要想对材料的自旋构型进行精确分析必须得到核外电子排布信息,这对材料表征技术的水平提出了挑战,因此自旋态的表征往往是自旋电催化中的难点。由于自旋与物质磁性的密切关系,磁化率曲线、电子顺磁共振等磁学表征可以在自旋表征中发挥关键作用。穆斯堡尔谱(Mössbauer Spectra)、X射线吸收光谱(X-ray Absorption Spectra,XAS)、X射线发射谱(X-ray Emission Spectra,XES)等最先进的光谱学表征技术能够提供材料电子结构信息,这为材料自旋的分析提供了机会(图10)。此外,随着计算机技术的迅速发展,模拟技术的精度已经达到或者超过了实验精度,基于密度泛函理论的和基于波函数的第一性原理计算能够从模拟计算的角度诠释材料的电子结构,解释反应机制,指导材料的设计。因此模拟计算技术也是自旋电催化研究中至关重要的一环。下面,本文将对电子自旋表征技术以及模拟计算技术进行详细介绍。

图10 电子自旋状态先进表征技术:X射线吸收谱图(XAS)、X射线发射谱图(XES)、振动样品磁强计(VSM)、穆斯堡尔光谱图(Mössbauer spectroscopy)、电子顺磁共振谱(EPR)Fig.10 Advanced characterization technology of electron spin-state: X-ray absorption spectroscopy (XAS),X-ray emission spectroscopy (XES),vibrating sample magnetometer (VSM),Mössbauer spectroscopy,electron paramagnetic resonance (EPR).
4.1 振动样品磁强计
振动样品磁强计(VSM)是测量材料磁性的重要表征技术,是一种基于电磁感应原理制成的高灵敏度磁矩测量仪器,它在探测线圈中振动所产生的感应电压与样品磁矩、振幅、振动频率成正比。在保证振幅、振动频率不变的基础上用锁相放大器测量这一电压,即可计算出待测样品的磁矩133。振动样品磁强计广泛用于测量各种铁磁、亚铁磁、反铁磁、顺磁和抗磁材料。表征技术可检测材料许多内禀特征,如磁化强度、居里温度等。由VSM可以测得磁化强度与磁场曲线(M–H)或磁矩与温度曲线(M–T),进而计算出材料的有效磁矩,在催化剂设计中可以通过有效磁矩计算得到活性中心原子未配对电子数,并推断出材料的自旋态134。
在电催化中可以利用VSM准确测定活性中心原子磁性已经自旋状态。Zhou等人在研究半无序策略调控催化剂自旋相关电子占据时,为了深入了解作为对称破缺的自旋相关电子占据,通过超导量子干涉装置在室温下测量了不同烧绿石的磁化强度与磁场(M–H)的函数关系。它们都显示出非线性磁滞回线曲线,显示出室温铁磁特性。根据Goodenough-Kanamori规则,铁磁特性主要由完全充满的eg能级和空的a1g轨道之间的电子跳跃决定。有序结构的烧绿石的饱和磁化强度明显高于无序结构的烧绿石和半无序结构的烧绿石,这可能与对称性破缺相关的自旋电子重构有关135。Zhang等人利用自行设计的电化学系统和可微调磁场强度(0–1.4 T)的振动样品磁强计,研究了原位外加磁场下镍基催化剂(Ni(OH)2、NiO和Ni)的析氧反应活性。磁滞回线表明样品在强磁场下具有室温铁磁或准铁磁行为,其中Ni具有53.83 emu∙g-1的最大饱和磁化强度。结果表明,自旋电子散射引起的磁电阻效应是影响磁场辅助析氧反应过程中表观电催化活性的主要因素,并且在第一电子转移步骤是OER的速率决定步骤的情况下,磁场诱导的自旋极化动力学将更加明显136。Gong等人利用交变磁场提高了单原子催化剂析氧反应活性,发现Co单原子在MoS2载体上的锚定(Co@MoS2)使其产生了面内室温铁磁性质,这有利于在施加磁场时氧原子的平行自旋排列。在M–H曲线中,Co@MoS2表现出明显的面内磁滞回线,表明其面内室温铁磁性和面外非磁性。随后在自旋轨道耦合计算中进行磁各向异性能的DFT计算,显示Co单原子在MoS2基面上的锚定不仅为OER提供了新的活性位,而且还具有面内室温铁磁性质137。
4.2 电子顺磁共振技术
电子顺磁共振(EPR)又可称为电子自旋共振(ESR),其利用具有未成对电子的物质在静磁场作用下对电磁波的共振吸收特性来进行表征分析,且EPR光谱只能分析含有未配对电子的顺磁或铁磁性物质138。常用作分析不同价态的过渡金属离子、烃或氧形成的自由基以及以有序自旋为特征的实体(如铁磁颗粒或导电聚合物)。通过对EPR光谱分析可以研究顺磁性物质的自旋-晶格弛豫、自旋-自旋耦合、自旋-轨道耦合等作用139。在能源催化中EPR技术常用于分析催化剂自旋态和配位情况,当样品中的电子自旋通过反铁磁相互作用耦合时,即自旋矢量反平行对准,导致总自旋为零时,EPR信号消失。但对于一些金属(如铁、钴和镍,以及一些氧化物如Fe3O4),它们是铁磁性的,因此样品中的所有自旋矢量平行排列,并且通常给予非常强的铁磁共振信号140。
Klasovsky等人研究了新型导电高分子负载金属催化剂(聚苯胺(PANI)负载的b-PtO2粒子)的氧化活性,发现催化剂能够在低温下将CO氧化为CO2,其利用EPR光谱显示纯PANI和PANI负载催化剂在空气中的EPR信号强度乘以温度I(T)∙T的曲线几乎平行,表明两催化剂具有相似的泡利磁化率值,PANI中极化子的浓度不受其表面上Pt沉积的显著影响。PtO2/PANI催化剂在393 K以上开始将CO氧化成CO2,并且同时I(T)∙T值也开始上升,表明源自CO氧化反应的电荷载流子通过强结合的PtO2物质转移到PANI载体的导带。由于纯PANI不发生这种效应,因此很可能强结合的PtO2对于促进这种转移是必需的。对此建立了EPR强度和催化剂活性之间的相互作用关系141。Wang等人利用W杂原子的充分掺杂调控Ni3S2纳米片(Ni3S2-W-Vs)中局域自旋态和能带结构提升了催化剂OER活性,其EPR光谱在g= 2.002处的特征峰归属于悬挂的Ni―S键证明了S空位的存在,Ni3S2-WVs比其他样品显示出明显更高的强度,表明峰强度与S空位的浓度有关。通过计算得出S空位的引入使d带中心上移,从而增强了*OOH的吸附142。
4.3 穆斯堡尔谱学
穆斯堡尔光谱学提供了一种研究局部配位结构、活性金属中心自旋状态(例如,Fe、Ni、Au、Ru、Ir等)的研究方法143。57Fe穆斯堡尔谱仪对铁的氧化态、电子自旋组态和配位环境具有很高的灵敏度,因此常用于探测铁的电子态和配位环境。通常会对穆斯堡尔谱拟合成为单重峰或具有对称性的多重峰(双峰、四重峰、五重峰、六重峰),依据每种峰的同分异构体位移(IS)和四极分裂(QS)数值结合实际情况分配Fe原子的自旋状态144。穆斯堡尔谱有三个重要参数与三种不同类型的相互作用有关。其中同分异构体位移是由电单极相互作用引起的,与金属原子核的4s电子密度有关,并间接(通过屏蔽效应)与3d电子密度有关。四极分裂ΔEQ是由铁核处的电场梯度(EFG,electric field gradient)引起的,表示了对立方对称性的偏离。除了同分异构体位移和四极分裂外,还存在磁相互作用的可能性,从而导致更复杂的光谱145。
在非贵金属催化剂中,Fe-N-C被认为是一种高效的氧电催化剂,57Fe穆斯堡尔光谱能够有效分析Fe原子自旋态,对于催化剂反应机理研究有重要作用。Li等人利用57Fe穆斯堡尔谱研究了S掺杂Fe-N-C催化剂的电子组态变化,将原始Fe-N-C的光谱归属于D1双峰,S改性Fe-N-C的光谱显示额外的D2双峰。D1和D2二重态峰属于具有中自旋态和高自旋态的FeN4物种。S掺杂后,FeII原子由中自旋态向高自旋态的转变使FeII原子更亲电,从而诱导O2的末端吸附。S改性Fe-N-C优化的未配对电子数和亲电性有助于优化氧物种与电催化剂之间的键强度,从而显示出更好的ORR活性。Liu等人分析了不同热解温度下FeNx中心自旋态、稳定性和活性,将拟合的四种双重峰定义为(D1–D4),并发现热解温度过高(800 °C)会产生单重峰的γ-Fe和六重峰的FexC,即Fe-N-C-600和Fe-N-C-700都只由Fe单原子组成,而Fe-N-C-800中存在Fe0粒子的聚集体。并且发现每种Fe物质的相对量取决于热解温度。D1 (FeIIN4)只存在于Fe-N-C-600样品中,表明这种结构在高温下不稳定。相比之下,D2 (高自旋XFeIIIN4-Y,(X,Y配体为O或N))存在于所有三个样品中。随着热解温度的升高,QS值降低,表明XFeIIIN4-Y结构变得更加对称。另一方面,D3 [低自旋N-(FeIIIN4)-N]的浓度随着热解温度的升高而降低,直到它在Fe-N-C-800中完全消失,在形成了γ-Fe和FexC的纳米颗粒。结果表明,由于在较高温度(800 °C或更高)下热解,D3结构被破坏,然后发生Fe0聚集。具有更多中自旋Fe-N5位的单原子Fe-NC催化剂,催化活性最高146。
需要注意的是,穆斯堡尔谱的应用存在着较大的局限。首先,具有穆斯堡尔谱效应的化学元素较为有限,能够充分进行穆斯堡尔谱测试的元素更为稀少,现今穆斯堡尔谱研究工作其中以57Fe为主(约占所有工作的75%),其次在过渡金属化合物中的长程结构和电子特性可能与催化剂活性位点不同,因此难以进行全面的结论性分析147。此外,穆斯堡尔谱测试中时间分辨率较低且M-N-C催化剂中M-Nx位点的低金属含量导致原位穆斯堡尔谱测量需要数十小时的光谱采集,在此期间样品的形态可能会发生显着变化。尽管穆斯堡尔谱技术的测试条件苛刻,其高分辨率,抗干扰能力强等优点依然使其在自旋表征方面具有极高的价值。
4.4 X射线谱学技术
由于磁学表征得到的自旋信息精确性不足,同时穆斯堡尔谱存在着一定的技术缺陷,自旋电催化的研究工作还需要其它的手段来分析材料的自旋情况。X射线谱学技术常常被用于分析物质的电子结构,可以作为自旋表征的重要工具。
4.4.1 X射线吸收光谱
X射线吸收光谱(XAS)常作为分析催化剂氧化态和配位环境所需的表征方法。当X射线透过样品后会发生一定的吸收和散射,并且其透射光强与原子质量相关,而XAS通过对比透过样品X射线强度和入射X射线强度关系,可以分析出样品的几何结构,元素化合价和元素自旋态等信息148。X射线照射样品产生的共振吸收使得电子电离为光电子,进而使X射线吸收系数发生突变,这种突跃称为吸收边(Edge)。通常根据其吸收能级将光谱分为两部分:吸收边前-吸收边后50 eV能级附近被称为X射线吸收近边缘结构(XANES),其吸收信号清晰,采谱时间短,适用于时间分辨实验,且对价态、未占据电子态等化学信息敏感,与之相对吸收边后50–1000 eV范围常被称为扩展X射线吸收精细结构谱(EXAFS),通过对其分析可以得到中心原子与配位原子的键长、配位数等信息149。
XAS在金属自旋态表征也具有显著的优势,在XANES中具有较低能量的前边缘峰的特征形状和分裂情况显示了金属的局部配体场和自旋状态。Hocking等人研究了两个化合物K3[Fe(CN)6]和K4[Fe(CN)6]的FeL-边的独特光谱特征,由于其表征了电子从2p→ 3d轨道跃迁并且这种转变显示出电偶极,这意味着FeL-边强度与未被占用的金属中的Fe 3d电子成正比。通过分析两种化合物FeL-边光谱,发现其特征是由于配体π*轨道与金属配体背结合而引起的,背结效应使光谱产生了更强的双峰强度特征。通过分析L边缘光谱形状、边缘能量位移和总强度,确定了FeL边XAS可以用来解耦和量化σ供给和π反供给在金属络合物中的影响150。Hu等人利用软X射线吸收光谱证明了层状Sr2CoO3Cl化合物中具有CoO5金字塔配位的Co3+离子明确处于高自旋态。实验中利用软X射线吸收光谱得到了化合物中CoL2,3边和OK边光谱信息,通过对比Sr2CoO3Cl化合物Co3+和Fe1-xO (x< 0.05)L2,3边以及EuCoO3光谱信息,发现样品与Fe1-xO光谱基本相同但与EuCoO3光谱有很大差异,由于Fe1-xO中Fe2+明确显示出高自旋态而EuCoO3中Co显示低自旋,通过对比其差异确定了样品中Co3+处于高自旋态。根据Harrison的公式,Co 3d到O 2p的电子转移积分适用于各种Co―O键长。这与晶场参数一起决定了Co3+离子是处于高自旋态还是低自旋态151。在OK边XAS光谱中发现从528到533 eV的结构是由于电子从O 1s能级到O 2p轨道的跃迁,混合到未被占据的Co 3d轨道t2g和eg态。随后对OK边XAS光谱进行了第一性原理计算发现Co处于HS态时的OK边谱可以用第一性原理计算很好地解释,从而佐证了Sr2CoO3Cl化合物中Co3+的高自旋态152。
4.4.2 X射线发射光谱
非共振X射线发射光谱(XES)不仅可以区分氧化和自旋态,还能通过结合能量色散设置在几分钟内进行采集的原位实验。
Saveleva等人利用XES量化了Fe-N-C催化剂的平均自旋状态。通过对铁卟啉(DW21)、金属有机骨架和醋酸铁混合物热解得到了Fe0.5催化剂,对两种催化剂(DW21和Fe0.5)进行了非原位XES测量,发现两种催化剂主峰Kβ1,3和伴随峰Kβ’位置基本一致,但其峰值强度不同。其中伴随峰是由Fe的3p和3d轨道交换作用产生,表示样品平均自旋状态(Saverage)。由于XES光谱受自旋状态和金属-配体共价双重因素决定,作者建立了XES光谱特征与结构相似化合物的自旋状态曲线,并分析了其相关性,发现Kβ’特征峰积分面积与自旋状态有明确的正向线性相关性,高自旋贡献导致更强的Kβ’特征。最后评估了DW21催化剂在聚合物电解质燃料电池应用相关条件下经历的自旋状态变化,发现随着电压变化,催化剂Kβ’峰积分面积随之变化,电位从OCV/0.9 V降至0.2 V,导致Saverage平均值从0.8降至0.55,并且在返回高电位时,发现这种变化逆转,证实了原位条件下催化剂的自旋状态受电势诱导变化153。
4.5 模拟计算方法
磁学分析中,模拟计算电子结构是一个非常重要的手段。其中经常采用的计算模拟方法是基于DFT的第一性原理计算。作为一种研究多电子体系电子结构的量子力学方法,第一性原理计算不仅在学术理论研究中快速发展,还在工业中逐渐得到应用。通过几十年的发展,计算机的计算能力和速度不断提高,使得密度泛函理论体系和其数值计算方法都有了很大的发展,现如今其在物理、化学、材料等领域得到了广泛应用154。DFT计算在能源催化中不仅可以计算反应过程热力学变化,还可以模拟计算材料晶体结构,以及催化剂电子能带结构和电子态密度,DFT计算技术的应用增进了研究人员对催化剂反应机理的研究155。
Wang等人研究了单原子W掺杂NiS0.5Se0.5纳米片的纳米棒结构催化剂(W-NiS0.5Se0.5),其具有高活性和高耐久性。通过计算发现,单原子W的引入使Ni的自旋态离域(图11a),导致Ni的d电子密度增加。这使得H的吸附/脱附过程得到优化,速率决定步骤(O* → OOH*)的吸附自由能显著降低,从而加速了HER和OER的热力学和动力学156。Xu等人通过波函数分析绘制了NixFe1-xOOH的自旋密度分布图,结果表明Fe位附近存在自旋通道,有利于局部自旋电流的传输(图11b),而Ni位上不存在自旋通道94。
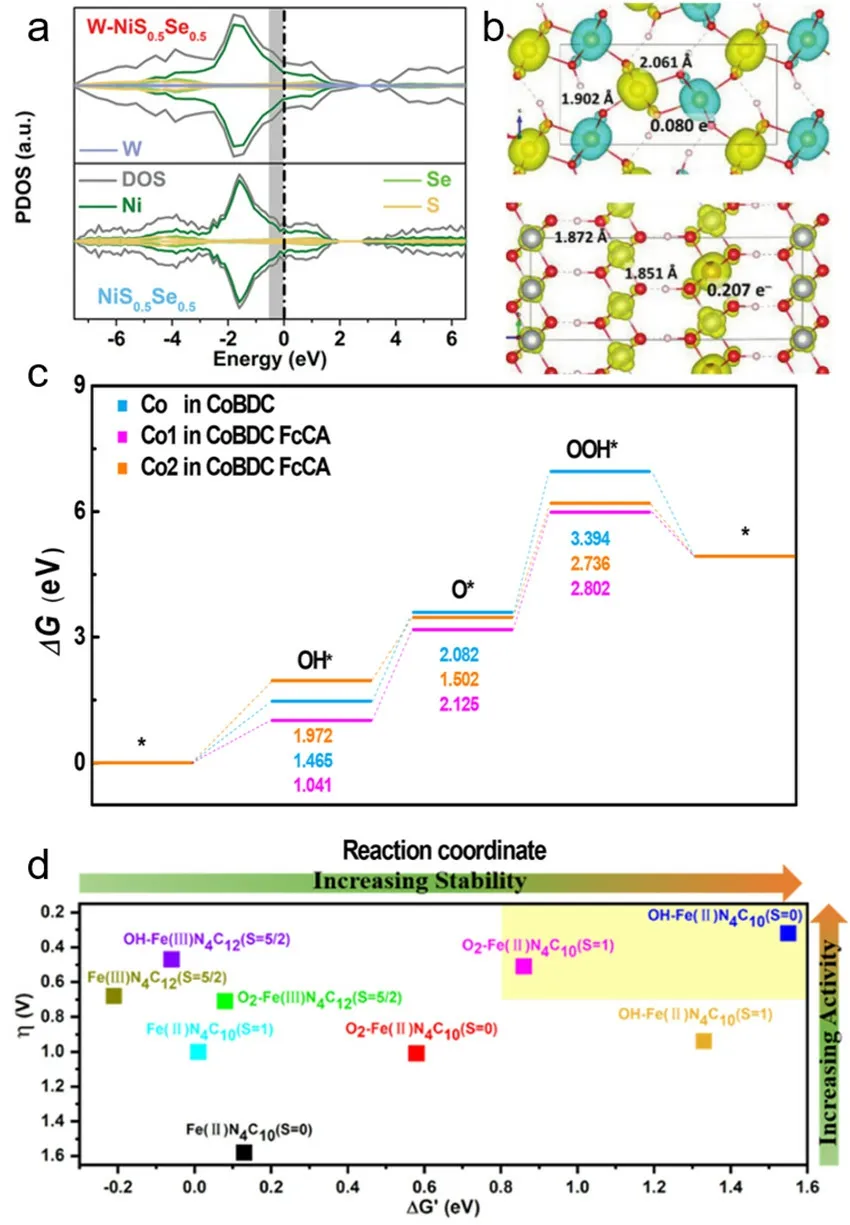
图11 模拟计算方法分析催化剂自旋。(a) W-NiS0.5Se0.5自旋极化投影态密度计算156。(b) NixFe1-xOOH自旋密度图94。(c) CoBDC FcCA的ORR台阶图157。(d)不同自旋态Fe-N-C催化剂的计算稳定性-活性关系图158Fig 11 Simulated calculation method to analyze catalyst spin.(a) W-NiS0.5Se0.5 spin-polarized projection density of states calculation 156.(b) NixFe1-xOOH spin density diagram 94.(c) ORR step diagram of CoBDC FcCA 157(d) Computational stability-activity relationship diagram of Fe-N-C catalysts with different-spin states 158.
He等人分析了双功能微反应器(CoBDC FcCA)通过电荷调制和应变在氧电催化中的活性,通过结构表征和DFT计算揭示了两种催化剂中Co活性基团与FcCA的配位作用,通过配位作用形成了一个张应变的微反应器,导致Co自旋从高自旋态(t52ge2g)向中自旋态(t62ge1g)转变,从而调节Co 3d和O 2p轨道的反键态。随后计算了催化剂在反应过程吸附能台阶图,发现CoBDC FcCA催化剂具有最佳的OH*吸附能,Co自旋态转变有利于OOH*中间体中O―O键的形成,从而有效地降低了速率控制步骤(O* → OOH*)的热力学吉布斯自由能,加快了整个OER过程的反应动力学(图11c)。CoBDC FcCA纳米片对OER表现出极好的催化活性,达到于10 mA∙cm-2的电流密度仅需280 mV的低过电位和53 mV∙dec-1的Tafel斜率157。
Tang等人计算了不同轴向配位官能团的Fe-N-C催化剂的催化活性和稳定性,计算结果表明低自旋的吡啶型FeN4[OH-Fe(II)N4C10(S= 0)]具有优异的耐酸性能和较高的ORR活性,而高自旋吡咯型OH-Fe(II)N4C10(S= 1)的ORR活性同样优异,然而稳定性较差(图11d)。这些结果为自旋调控的电催化剂的活性和稳定性设计提供了丰富理论指导158。
5 结论和展望
自旋调控作为研究电催化活性机制的一个新兴方向,可通过调控活性中心的自旋态和催化剂的自旋极化来优化催化反应中的电荷传输进而调节活性中心与反应物/中间体的吸附作用,自旋效应的利用为高效电催化剂的开发提供了一种新的设计策略。本文综述了自旋相关电催化的最新理论和实验研究进展。我们首先介绍了电子自旋以及自旋的调控方式,随后总结了自旋效应在电催化中可能的理论机制,并介绍了如量子自旋交换相互作用,自旋磁熵等重要概念。进一步,我们整理了自旋效应在ORR、OER、CO2RR、NRR等反应中的应用并分析了可能的机制,最后归纳了电催化剂自旋表征技术和DFT计算进展。随着电催化材料自旋效应的深入研究,结合多种先进表征技术以及理论模拟计算以及对电催化剂自旋调控的开发,自旋效应有望推动电催化剂的发展。需要指出的是,由于研究历史尚短,自旋效应在电催化剂设计中的应用还存在很多挑战性问题亟须研究。下面,本文提出未来自旋电子学在电催化中的可能发展方向(图12)。
图12 电催化剂设计中的总结与展望Fig.12 Challenges and perspectives for spin effect in electrocatalysis.
(1)活性位点自旋态的精准调控策略。尽管目前一些报道通过改变活性位点的自旋态实现了提升催化剂活性的目标,然而这些材料存在多种价态与自旋态活性位点共存的问题,无法实现对催化剂自旋的精确调控,难以得到自旋电催化研究的理想材料。以调节材料磁学性能为目的自旋调控已有大量研究,结合自旋电子学的研究经验可能有助于实现电催化剂自旋的精准调控。需要注意的是用于先进电催化剂的材料研究方向与自旋电子学研究的主流材料存在差异,必须结合两者特点谨慎理性进行电催化的设计。
(2)人工智能结合高通量实验平台促进材料开发的新模式。对于电催化剂的自旋理解推测离不开DFT计算的指导。然而单纯的DFT计算方法效率低下、容易浪费大量资源。人工智能是一种新型、快速、高效的电催化工作参数优化工具。利用机器学习结合第一性原理计算将会产生新的材料预测和设计方法,人工智能可用于识别自旋效应描述符,以合理预测催化活性,加快新型自旋电催化材料的设计,并推导出催化反应中的反应机理与活性中心的自旋性质的关系。同时必须将DFT研究与实验验证相结合,高通量实验平台有能力对机器学习得到的大量数据批量快速进行实验验证。这些新工具能克服传统的实验为主的“试错法”的时间成本问题,对预测高效自旋催化剂的性能和指导高效自旋催化剂的研究具有重要影响。
(3)探索自旋效应在更多催化反应中的应用。自旋效应对电催化反应的促进作用已经在ORR、OER、NRR、CO2RR的研究中得到验证,并为高效电催化剂的研究提供了一个新的方向。自旋作为粒子的内禀性质,有可能影响许多反应的进行。热催化在石油加工、化学工业和制药工业中起到重要的作用。在有机催化合成中的,手性分子合成过程中电子自旋产生了独特作用,但在其他有机合成反应中活性中心的过渡金属也有可能通过电子自旋效应影响催化活性。因此我们认为自旋效应的深入研究可以拓展到热催化领域中,为催化剂设计带来新的思路。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Introducing Novel,Multiple Cd Coordination Modes into Gold Nanoclusters by Combined Doping for Enhancing Electrocatalytic Performance
- 用于高灵敏快速核酸检测的荧光碳点
- 利用多氟丙烯酸酯添加剂提升准二维钙钛矿发光二极管性能
- Recent Advances in Self-Supported Transition-Metal-Based Electrocatalysts for Seawater Oxidation
- 高效光电调控钙钛矿量子点阻变存储性能
- 电催化二氧化碳还原催化剂、电解液、反应器和隔膜研究进展