电催化二氧化碳还原催化剂、电解液、反应器和隔膜研究进展
2024-01-22彭芦苇张杨何瑞楠徐能能乔锦丽
彭芦苇,张杨,何瑞楠,徐能能,乔锦丽,2,*
1东华大学环境科学与工程学院,纤维材料改性国家重点实验室,上海 201620
2上海市污染控制与生态安全研究院,上海 200092
3香港理工大学应用物理系,香港 999077
1 引言
人类社会的正常运转需要化石能源的供给,而化石能源的消耗导致了能源的紧缺和环境污染,但人类社会对化石能源的依赖却日益增加,从全球能源消费结构中可以看出,煤炭的占比为27.2%、石油的消耗占比约为33.62%、天然气的占比约为23.86%1。由于化石能源的存储量有限且人类对化石能源开采仍在加剧,因此人类社会正在面临严峻的能源危机。将化石燃料作为能源供给体,也导致CO2排放量急剧增加,根据Carbon Monitor全球实时碳数据显示,全球每天的碳排放量在100 Mt (百万吨)浮动(图1a),我国每天的碳排放量在32 Mt左右,约占全球总排放量的32%(图1b),其中燃煤发电、水泥和钢铁行业烟道气中排放的CO2占我国总排放量的90%2。据美国National Oceanic and Atmospheric Admanistration(NOAA)全球检测实验的数据,全球平均二氧化碳浓度在2021年创下新高:414.72 × 10-6(图1c)3。二氧化碳在地球系统中很重要的原因是,它就像溶解在苏打水中一样,可溶解在海洋中,还可与水分子发生反应,产生碳酸并降低海洋的pH值(提高海水的酸度)。自工业革命以来,海水(海洋表层)的pH值从8.21下降到8.10,这种pH值的下降称为海洋酸化,会导致海洋生物的大量灭绝4。CO2气体分子又称为“温室气体”,因为它对来自太阳辐射的可见光具有高度穿透性,而对地球发射出来的长波辐射具有高度吸收性,能强烈吸收地面辐射中的红外线,导致地球温度上升5,从图1d全球地表温度的变化可知,除1998年外,最热的年份中有十九年发生在2000年以来,2020年与2016年并列,是自1880年开始记录以来最热的两年。全球变暖引发最直接的环境问题是:冰川和冻土消融,冰川消融已经导致海平面升高,这使得很多沿海城市面临被淹没的风险。科研人员从极地钻取的冰芯中发现,冰芯中的古老病毒,即使经过了几千万年的演化,仍然是活的,因此冰川消融还将导致大型流行病毒的传播,尚不确定新型冠状病毒(Covid-19)的传播和冰川消融有关。

图1 (a)全球碳排放图2;(b)中国碳排放图2;(c) 1960–2021年空气中CO2的月平均浓度3;(d)全球从1880–2020年球地表温度的变化图3Fig.1 (a) Global carbon emission map 2; (b) China’s carbon emission map 2; (c) monthly mean concentrations of CO2 in air from 1960 to 2021 3; (d) map of global change in global surface temperature from 1880–2020 3.
2020年9月,中国在联合国一般性辩论谈话中向世界庄严承诺,中国将提高国家自主贡献力度,采取更加有利的政策和措施,二氧化碳排放力争于2030年前达到峰值,努力争取2060年前实现碳中和6。虽然CO2在空气中的大量排放将导致一系列环境问题,但CO2作为廉价的C1原料,可通过催化反应转化为高附加值的燃料和化学品,不仅为我国节能减排做出巨大贡献,还可开辟一种燃料合成新路径7。在国家宏观政策的指导下,CO2节能减排极大地刺激着科研从业者和企业家们的兴趣,并着手研发一些CO2减排技术,最简单和直接的方法是植树造林,然而植物只能吸收和转化空气中的CO2,因此人工造林仅对分布式碳捕获有良好的效果,这种方法对水和土地的要求并不适用于集中碳排放的情形(如火力发电厂和工厂),取而代之的应该是一些更加经济可行的方法。如上所述,诸如人工造林之类的低成本方案虽然不适用于二氧化碳集中排放的场所,但其所依靠的光合作用原理对于如何集中利用二氧化碳有一定的借鉴意义8。例如,自然状态下的光合作用是将水作为质子来源,并产生氧气作为副产物的过程,通过设计类似的反应,在确保不破坏全球生态的条件下,使其比自然界的光合作用更加高效,这将非常具有应用前景。
利用可再生能源作为驱动力,水作为质子和电子供体,将CO2转化为碳基燃料或化学品的主要措施有:1)光催化法;2)生物化学转化法;3)热催化法以及4)电化学转化法,如图2所示9。光化学转化CO2与光合作用过程类似,其本质是半导体光催化剂在不同波长的光照下产生光生电子-空穴对的过程,同时在半导体上发生氧化还原反应,即H2O的氧化和CO2的还原,但该方法需要光辐射的能量高于半导体的禁带宽度,电子才会从半导体价带跃迁到导带,且光生电子-空穴还有恢复到原状态的可能性,导致光化学转化CO2的效率较低10。电力驱动的微生物CO2转化法则是一个多步的过程,参与电合成的微生物可直接作为电极,用于提供电子并驱动胞内代谢反应将CO2还原为甲烷、乙酸和乙醇等有机化学制品,但该技术的主要瓶颈为:1)微生物与电极间的胞外电子传递速率较低,导致反应速率较慢;2)产品的附加值低11。热化学转化CO2顾名思义即在高温条件下将CO2转化各种化学品,如甲醇、乙醇、烯烃等12,应用于该方法的催化剂必须在高温条件下保持较高的活性,如何增加CO2的转化率和选择性将是该领域未来工业化进程中的研究重点,且尾气的再利用也应该引起重视。在众多转化技术中,CO2电还原(CO2RR)技术由于条件温和、反应可控、对环境友好和产物众多(甲酸、甲醇、一氧化碳、乙醇和乙烯等)受到广泛关注,但电催化转化CO2往往伴随着竞争反应-氢气析出(HER),如何抑制氢气的析出反应,使CO2RR具有优异的选择性、活性、稳定性是该领域着重考虑的三个因素13。

图2 (a)光化学和光电化学示意图;(b)生物化学法示意图;(c)热化学法示意图;(d)电化学法示意图Fig.2 A schematic depicting selected (a) photochemical and photoelectrochemical; (b) biochemical;(c) thermochemical and (d) electrochemical methods.
2 CO2电催化还原机理
CO2RR反应在不同催化剂和工况下将进行多质子-电子偶联过程,反应路径是2e-、4e-、6e-、8e-、12e-或更高电子数参与的反应,不同的反应路径对应着不同的还原产物,即使参与反应的电子数相同,产物也千差万别。因此CO2RR的主要产物包括一氧化碳(CO)、甲酸(HCOOH)或甲酸盐(HCOO-)、草酸(H2C2O4)或草酸根(C2O42-)、甲烷(CH4)、甲醛(CH2O)、甲醇(CH3OH)、乙烯(C2H4)、乙醇(CH3CH2OH)等,见表114和图315。CO2RR两电子反应有气体产物CO和液体产物甲酸HCOOH或甲酸盐,无论是产CO路径还是产甲酸盐路径都需要经过最初的CO2活化形成CO2*自由基(*代表了活性位点),如公式(1),(2)。CO2*自由基在催化剂表面的构型决定了不同的反应路径,如CO2*自由基在金属-碳-氮或金属-碳-氧键上倾向于产生*COOH (产CO的中间体,公式(3)),之后*COOH再结合一个质子和电子产生CO的前驱体(*CO,公式(4)),最后*CO从电极表面脱附形成气体产物CO(公式(5))。液体产物甲酸或甲酸盐路径和产CO路径的不同发生在形成CO2*自由基之后,即发生质子偶合电子转移的过程形成*OCHO-(甲酸盐的中间体,公式(6)),之后*OCHO-中间体从电极表面脱附形成甲酸盐(公式(7)),公式(1)–(7)的内容可形象地从图3中看出。

表1 在标准条件下(1.0大气压和25 °C)CO2转化为各种C1和C2产物的标准电位14Table 1 Standard hydrogen electrode (SHE) for electrochemical conversion of CO2 to various C1 and C2 products under standard conditions(1.0 atmosphere and 25 °C) 14.
除了发生2e-转移产CO和甲酸盐外,CO2RR常见产物还有发生6e-转移的液体产物,如甲醇。图3中产甲醇的反应路径为:CO2→ COOH* → CO* →COH* → CHOH* → CH2OH* → CH3OH16,当CO2被吸附在电极表面(通常铜(111)面)时,CO2中的一个O将同时与H2O中的一个H成键,并得到一个电子形成COOH*中间体,在这个质子-电子参与的过程中,通常也发生着C―OH键的断裂,即COOH*中间体脱水形成CO*自由基的过程,水脱附后CO*自由基在电极表面的脱附远远难于CO*加H形成COH*自由基,需要注意的是CO*加H可形成CHO*或COH*,但CO*还原成CHO*从热力学角度上所需要吸收的热远远高于还原成COH*,因此COH*更容易形成。接下来是COH*的还原,H2O中的一个H与COH*中的C成键形成CHOH*中间体,COH*形成CHOH*中间体的过程是整个反应中吸热最多的,也是整个反应的决速步骤,CHOH*还原到CH2OH*也是H2O中的一个H和中间体中的C成键,最后CH2OH*中间体从电极表面脱附形成液体产物CH3OH。Mou等17进行了CH2O电化学还原实验证实了*OCH2是CO2还原到CH3OH的中间体,如图4a。上述讨论过的三种产物(CO,HCOOH,CH3OH)都是CO2还原的C1产物,另一个常见的C1产物是气体产物CH4,产甲烷的反应路径为:CO2+ * → COOH* → CO* + H2O → COH* →CHOH*→ *CH + H2O → *CH2→ *CH3→ * + CH4,如图3和图4b18,同样在Cu(111)面上,产CH4路径与产CH3OH路径从CO2到CHOH*中间体是相同的,不同的是CHOH*中间体之后的反应,产CH3OH路径中,CHOH*中间体之后是偶联质子的过程,而产CH4路径中,CHOH*中间体之后是脱水的过程,即CHOH*脱水形成*CH和H2O,然后*CH连续偶联质子最后生成CH4离开电极表面19。
除了C1产物外,C2H4和CH3CH2OH是最常见的C2产物,尽管CO2到C2反应路径复杂,但CO2首先被还原为CO*,然后再进行C=C偶联反应以及后续的加氢加氧过程,如图3。目前CO2电化学还原到C2H4的机理一直在研究中,在不同催化剂表面反应中间体也不尽相同。如Qiu等20用八羟基酞菁铜和CuO4作为催化剂研究了CO2到C2H4的机理,提出产C2H4的反应路径为:CO2+ * →*COOH → *CO → *CHO + CO → *COCHO →*COCHOH → *CHOCHOH → *CCHOH →*CCH → *CCH2→ *CHCH2→ *CH2CH2→ * +C2H4,如图4c。从反应路径可以看出,C2H4的产生需要*CO和质子化的*CHO吸附在电极表面并发生C=C偶联反应,八羟基酞菁铜对*CO的吸附能(48 kJ∙mol-1)远高于CuO4(16 kJ∙mol-1),故在八羟基酞菁铜上更易发生加氢生成*CHO,另外CuO4将CO2转化为*CO具有较高的活性,因此CuO4可为C=C偶联过程提供*CO,需要注意的是,*CO需要先从CuO4上脱附才可参与C=C偶联反应生成*COCHO,C=C偶联反应后进行的是加氢过程,即陆续在两个C上加氢生成*COCHOH和*CHOCHOH,接下来的过程是在两个C上陆续发生脱氧的过程生成*CCHOH和*CCH,然后分别在C上加氢直至生成C2H4。Meng等21在Cu(200)面上研究了CO2到C2H4的反应路径:CO2+ * → *COOH→ *CO → *CO + *CO → *OCCO → *OCCOH →*CCO → *CHCO → *CHCHO → *CH2CHO → *O +C2H4,如图3和图4d,从反应路径上可知,在Cu(200)面上*CO与*CO之间直接发生C=C偶联反应生成*OCCO,值得注意的是C=C偶联反应是整个反应的决速步骤,然后其中一个C与水中的H成键形成*OCCOH,与此同时发生的反应还有C―OH键的断裂,即*CCO的形成,随后*CCO得到一个质子和电子生成*CHCO,再连续得到质子和电子分别生成*CHCHO和CH2CHO,最后再得到一个质子和电子脱离*O生成C2H4。上述两条C2H4路径的不同之处在于C=C偶联反应和偶联后的反应,因此针对不同的催化活性表面,C2H4是否有普适性的反应路径仍将是整个领域的研究重点。
考虑到C2H4和C2H5OH是所有C2产物中选择性最有可能超过50%的产物,研究C2H4和C2H5OH反应路径的分歧点对提高单一产物的选择性具有指导意义,根据C2H4的反应路径可知,CO2首先被还原为*CO才可进行C=C偶联反应,因此,Koper等22在Cu(100)面上用CO来研究C2H4和C2H5OH反应路径的不同(图4e),当CO分子吸附在Cu(100)面后首先形成*CO,之后将进行C=C偶联反应形成*OCCO中间体,这和Meng等21在Cu(200)面上的研究结果相一致,不同的是C=C偶联后的反应,Koper等22认为在*OCCO中间体上加氢有两种方式,分别为H与其中的一个C成键生成*COCHO,或H和其中的一个O成键生成*COCOH,由于*COCOH中间体比*COCHO更稳定,故*OCCO中间体的加H更倾向于发生在O上,*COCOH中间体可以被加氢生成*OHCCOH、*OHCCHO和*OHCHCO中间体,也可以脱水形成*CCO + H2O,根据结构化学和还原态条件,*COCOH中间体中的OH基团相比于羰基(C=O)基团中的O更易质子化和随后的脱水,故*COCOH中间体会发生C―OH的断裂生成*CCO,之后*CCO连续分别在两个C上偶联H生成*CHCO、*CHCHO和*CH2CHO,产C2H4和C2H5OH的区别在于*CH2CHO中间体之后的反应。C2H4路径接下来的质子偶联电子发生在与O相连的C上,即*CH2CHO分子式中后边的C与H结合,然后C=O键断裂生成*O和C2H4,C2H4从电极表面脱附形成气体产物,之后*O连续参与质子偶联过程生成*OH和H2O。产C2H5OH的路径在*CH2CHO中间体之后同样发生的是质子偶联电子的过程,但H是与*CH2CHO分子式中前面的C结合,生成*CH3CHO,再紧跟着两步质子偶联电子过程生成*CH3CH2O和CH3CH2OH,故H3CH2OH的反应路径可总结为:CO2+ * → *COOH → *CO → *CO + *CO →*OCCO → *CCO → *CHCO → *CHCHO →*CH2CHO → *CH3CHO → *CH3CH2O →CH3CH2OH,如图3和4e。由此可知CH3CH2OH路径和C2H4路径在*CH2CHO之前的反应是一样的。通常在铜基催化剂上,C2H4的选择性高于CH3CH2OH,这主要因为在*CH2CHO中间体上脱O比在其上加H具有较低的能量。
3 CO2RR催化剂研究进展
CO2RR是在负的电极极化下和大量质子存在的情况下发生的,1994年Hori等23在不同的高纯金属表面上研究了CO2的还原产物,并根据还原产物的不同,将金属分为四类:产氢、产CO、产甲酸以及产多碳化合物。在0.1 mol∙L-1KHCO3作为阴极电解液时,金属Ni、Fe、Pt、Ti的还原产物是H2,CO2在这些金属表面并不能被还原;过渡金属Pb、In、Sn、Hg、Cd、Bi的还原产物是甲酸盐;金属Au、Ag、Zn、Pd、Ga的还原产物主要是CO;铜金属具有优异的CO2还原能力,还原产物主要有CO、HCOOH、CH4、C2H4、CH3CH2OH,以及其他多碳产物24。随着CO2RR领域研究的深入,各种金属和非金属催化剂也得到了突飞猛进的发展,因此有必要根据产物的不同做简要的归类,由于C1产物(CO和甲酸)最具有商业化的前景,且C2产物(C2H4和CH3CH2OH)的主要催化剂以铜基为主25,最近研究发现,Ni作为催化剂也能够将CO2还原为碳氢化合物,但需要在高过电位下,且法拉第效率非常低26。此外,CO与Ni的强结合会导致Ni表面中毒,将Ni做成单原子或与其他金属合金化,可以有效地破坏Ni与CO的吸附27。尽管目前研究发现Ni可作为电催化CO2还原为碳氢化合物的催化剂,但被大家普遍认同的,仍以Cu基催化剂为主。以下将简要介绍产CO和甲酸的催化剂。
3.1 产CO的催化剂
CO2RR产物中只有CO和HCOOH的法拉第效率(FE)可以高达90%以上,乙烯的FE最高不超过80%,乙醇的FE更是不超过55%,尽管C2产物相比于CO具有较大的市场规模和能量密度,但C2产物往往涉及多电子和多质子转移过程,从耗电和反应器材料费等角度来看,CO和HCOOH是最有可能商业化的产物。甲酸是液体产物且溶于电解质中,因此甲酸的利用往往需要分离和提纯过程,造成额外的技术成本。产CO路径的另外一个优势在于,CO2RR的气体产物只有CO和H2(竞争反应),因此可通过改变实验条件实现CO和H2比例的调控,使其满足工业上的费托合成反应,来生产多碳化合物。虽然CO2RR产CO在工业上已展现出独特的优势,但其产物气体和CO2气体的分离仍会导致成本的上涨。如何合理的调控CO和H2的比例以及在大电流密度下实现CO的规模化生产,核心的问题便是催化剂的开发,从纳米科学的角度思考,有效活性位点的数量和微环境下的电子结构将直接影响催化剂的选择性和活性,根据不同催化剂类型可归纳如下。
3.1.1 金属基催化剂
Kanan等28用恒电位电化学氧化法制备了Au纳米颗粒的氧化物电极,然后在负的电势下还原成Au金属电极并用于CO2RR产CO,发现在过电势仅为140 mV的情况下CO的FE可高达95%,且可稳定工作8 h,通过将纳米Au金属电极与多晶Au和其他结构的Au电极做对比,发现多晶Au电极只有当过电势大于200 mV时,电极才表现出比较好的CO2RR活性,但性能衰减的很快,因此催化剂的微观结构对CO2RR性能具有直接的影响,经过电化学动力学研究发现,经过电化学氧化和还原后,纳米Au金属表面对于CO2*-的吸附得到了显著提高,因此可调控催化剂的微观结构实现对CO2*-中间体的吸附能力,进而提高产CO的速率和降低反应的过电势。Liu等29利用电沉积法合成了不同形貌的金纳米针、金纳米颗粒、金纳米棒,并对它们进行了CO2RR性能测试,发现在-0.3 Vvs.RHE (相对于可逆氢电极)时金纳米针的电流密度可达7 mA∙cm-2且CO的FE可达90%,而金纳米颗粒的电流密度仅为0.05 mA∙cm-2且伴随着强烈的析氢反应,金纳米棒的电流密度仅为0.1 mA∙cm-2且CO的FE仅为3%,针对形貌的不同造成性能巨大的差异,有限元数值模拟分析揭示了纳米尖端处的电场强度是平面的十倍之多,因此电解液中的K+会在纳米尖端处聚集,根据第一性原理(DFT)模拟可知,K+在纳米尖端的吸附会导致CO2到COOH*中间体自由能的降低(图5),因此,碱金属离子在活性位点的聚集可调控CO2RR的性能。除了活性位点附近的微环境会改善CO2到中间体的吸附能,活性位点的位置和数量也同样会影响CO2RR反应。Sun等30用简易的种子介导生长法合成了超薄金纳米线,发现在金纳米线上平均配位数是6,而在金的(211)面上平均配位数为7,在金角落处配位数是5,由于配位数越小代表结合能力更强,因此金纳米线结合COOH*中间体的能力比金平面强,而金纳米线对CO的结合能力却弱于金角落处,通过DFT模拟分析也可证实这一点,金纳米线较强结合COOH*中间体的能力非常有利于COOH*脱水到CO,而弱结合CO的能力又非常有利于CO的快速脱附,故在-0.35 mA∙cm-2时金纳米线可将CO2还原到CO的FE可高达94%。Smith等31利用电化学氧化的方法先将Ag电极氧化,然后氧化的电极将在CO2RR的过程中被重新还原到单质状态,发现经过电氧化后Ag电极CO的FE可达80%,经电极动力学分析可知主要是由于氧化后的电极结合COOH*中间体的能力得到了增强,且在CO2RR进行的过程中伴随着质子的偶联反应,因此Ag纳米结构处的微环境pH将升高,高的pH将抑制析氢反应。2015年Rosen等32为了验证Ag纳米结构的上述优点,对比了Ag片、纳米颗粒、纳米多孔结构的CO2RR性能,首先DFT计算得出了,在Ag纳米多孔结构上反应速率是Ag平面的6倍,此外第二个质子参与的反应是CO2到CO的决定性步骤,最后提出了一种CO2RR的反应机理,即需要从动力学、CO2分压力和HCO3-的浓度等综合因素考虑CO2RR反应。从催化剂的微观结构出发,Lu等33利用去合金法制备了纳米多孔Ag催化剂,在过电势不超过0.5 V时,CO的FE为92%,这主要因为纳米多孔结构可为CO2的还原提供不同深度和高曲面的活性位点,进而CO2*的结合将被加强,故而表现出如此优异的活性。
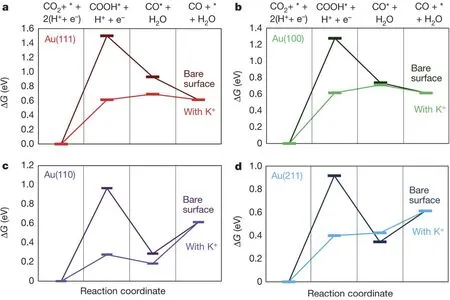
图5 电化学还原CO2到CO的吉布斯自由能图29Fig.5 Gibbs free energy ΔG diagrams of the electrochemical reduction of CO2 to CO 29.
为了更加清晰地理解Au和Ag在CO2RR反应中的不同之处,Back等34研究了Au和Ag纳米颗粒的活性位点对CO2到CO反应的影响,对于纳米颗粒而言,低指数晶面、边缘位置和角落位置往往认为是CO2RR反应的活性位点,Au的所有低指数晶面对于*COOH的结合都要强于Ag,而低指数位点对于中间体的结合能要比低指数晶面更强,这也可证实为何多孔结构具有优异的CO2RR性能,由于(111)晶面是所有低指数晶面最稳定的,从Au和Ag的(111)晶面思考,发现在Ag的面、边、角位点对*COOH的自由能分别为1.25、0.79、0.95 eV,这说明Ag边对于CO2RR的活性最高,这与Lu等的研究结合保持一致,而在Au上仅为1.21、0.70、0.62 eV,说明了金角对CO2RR的活性更高,这与Sargent和Sun的研究结果相一致。具有更强*COOH的结合是CO2RR到CO具有优异活性的基础,但同样在活性位点处也要考虑对于*H的吸附,因为较强的*H结合意味着HER的加强,尽管从*COOH的自由能发现,Au的活性要优于Ag,但Au对于*H的结合能要强于Ag,说明了HER更易在Au发生,无论是Au还是Ag,发生HER的顺序是边、面、角位点。另一点需要注意的是,由于CO2RR到CO是需要质子参与的反应,而竞争反应也是氢气的析出反应,因此在活性位点处将产生大量的*OH,*OH在活性位点的覆盖需要尽快清除以妨碍CO2RR的进行或防止催化剂的过氧化,值得注意的是Au对于*OH的结合要弱于Ag,这说明了在CO2RR进行中,Au不易被氧化故而活性优于Ag (图6)。

图6 *OH脱附反应随EB[*OH]变化的自由能图34Fig.6 Free-energy changes for *OH removal reaction are plotted vs.EB[*OH] 34.
单一金属基CO催化剂以贵金属为主,而双金属基催化剂可通过电子结构效应、应力效应和几何效应来提高CO2到CO的活性、稳定性和选择性。Hao等35利用静电纺丝法合成了均相纳米AuNi合金,发现该合金催化剂在-0.98 Vvs.RHE时CO的选择性可达92%,CO2RR性能优异的原因是Au相比于Ni具有更大的电负性,导致Au和Ni在合金中有很强的电子效应,DFT模拟也证实了将Ni引入Au后可使d-band中心变得更正且可降低CO2活化成*COOH的活化能,也可降低*CO的脱附能,从而避免Au催化剂CO中毒。Dai等36通过将Ni(OH)2纳米片中的Ni2+部分替换成Cu2+后,再将表面的Cu2+还原成Cu(0),形成的铜层只有原子层厚度且具有非常好的抗氧化性能,在CO2RR过程中,生长于Ni(OH)2纳米片上的铜层可表现出类似Ag和Au的催化性质,在-1 Vvs.RHE时,CO的法拉第效率可达92%,电流密度可达4.3 mA∙cm-2。Woyessa等37利用g-C3N4作为载体在其上负载Cu2O-FeO纳米颗粒,发现基底与Cu2O-FeO之间的接触非常紧密且有电子转移效应,因此该复合催化剂的CO选择性为96%,电流密度为4.65 mA∙cm-2。Zeng等38首先采用电沉积法在铜片上合成三维多孔的铜电极,然后在含锡前驱体溶液中二次电沉积锡用于改性铜电极,发现复合电极具有核壳结构,其中核是金属铜,壳是CuOx/SnOx物种,该复合电极在-0.75 – -0.9 Vvs.RHE的电势窗口下CO的法拉第效率可达93%–94%,CO的分电流密度为4.7–7.9 mA∙cm-2。基于核壳的概念,Li等39在Cu纳米颗粒表面涂敷一层厚度仅为0.8 nm的SnO2层,发现Cu原子可扩散入SnO2层,从而降低COOH*的形成能,因此CO是CO2还原的主要产物;而SnO2层的厚度为1.8 nm时,Cu原子并不能扩散到SnO2的外层,CO2还原的主要产物是甲酸。Ju等40也发现在Cu修饰的聚偏氟乙烯纳米纤维上Sn的厚度也会改变CO2还原的选择性,当Sn/Cu层的厚度为47 nm时,CO2还原到CO的法拉第效率可达80%,但Sn厚度过量时,主要产物是甲酸。
3.1.2 单原子催化剂
对块体催化剂而言,不可避免的是活性位点的聚集,这导致了活性位点的利用效率低,单原子催化剂可以很好地解决上述问题,几乎最大化的原子化效率和不饱和配位环境不仅降低了金属的大量使用,也增强了催化剂的催化活性41。通常单原子负载于导电基底上,目前广泛使用的基底包括碳材料(石墨烯、碳纳米管、无定形碳、C3N4、多孔碳等)和金属氧化物(TiO2、CeO2、In2O3、和ZrO2)等,负载的单金属主要为Ni、Fe、Co、Cu和Zn。负载的单原子金属稳固在基底上是通过与其化学成键,通常负载金属是和基底上的缺陷成键或配位,配位的原子一般为N、S、P、O等,因此单原子周围的配位原子和配位数将直接影响CO2RR的性能,降低单原子金属原子的配位数通常被认为可提高金属原子对CO2和中间体的吸附42。Ni单原子是CO2RR到CO研究最多的催化剂,Li等43用局部化学转换策略将Ni-N4稳固在g-C3N4上,Ni-N4活性位点相比于N-C可降低COOH*的形成能,因此CO更易在Ni-N4上形成,所以CO的FE在电势区间内(-0.5 – -0.9 Vvs.RHE)可达90%,而在-0.81 Vvs.RHE时CO的FE为99%且电流密度为28.6 mA∙cm-2。黄小雄等44通过热解含镍金属有机框架结构得到了负载高含量镍单原子(7.77%,质量分数)的超薄氮掺杂二维碳纳米片,在热解过程中会发生结构转变,即Ni2+-N-C转变成Ni+-N-C,Ni+-N-C具有较大的比表面积和多孔结构,并可作为CO2还原为CO的活性位点,因此在-0.77 Vvs.RHE时,CO的法拉第效率可达到99%,电流密度为12.6 mA∙cm-2。由于氧化石墨烯表面和边缘存在含氧官能团(如环氧、羟基、羧基、羰基),通过静电相互作用对水溶液中的金属阳离子具有较高的吸附能力,因此氧化石墨烯作为基底可负载Fe单原子,再将氧化石墨烯和FeCl3在Ar/NH3气氛下650–800 °C煅烧,即可实现对氧化石墨烯的还原和氮掺杂,在石墨烯中引入氮原子可用于固定Fe原子,通过改变FeCl3的添加量可实现Fe以单原子、纳米团簇、纳米颗粒等形式负载于石墨烯上,其中Fe以单原子的形式存在时,CO2RR到CO的性能最佳,这主要是氮原子和原子级别Fe的相互作用,而Fe的配位环境主要是氮,Fe-N4的存在更有利于CO2转化为COOH*中间体45。
为了探究金属位点对CO2RR的影响,Pan等46分别将Fe、Co、Ni、Cu以单原子的形式负载在中空碳球上,并证实了Co位点相比于Fe、Ni、Cu具有更加优异的产CO性能,这主要因为Co与5个氮原子配位形成Co-N5结构,因此Co-N5作为活性中心可将CO2快速转化为COOH*,且Co-N5结构更有利于CO的脱附。由于金属催化剂和金属氮碳材料(M-N-C)对CO2还原的差异,研究它们的限制性步骤对设计催化剂具有指导意义,CO2到CO有两个电子转移,CO2得到一个电子变成CO2-在金属电极上不可能是决速步骤,主要因为这一步骤的限制来源于溶剂重组(CO2-到CO3-)和电子从CO2*转移到CO2-的速度,当电子从电极表面转移到吸附CO2*的s和p轨道上的速度相比于CO2*从溶剂中扩散到电极表面的速度很小时,电子转移不可能是决速步骤,可考虑CO2*的吸附和COOH*的形成是否是决速步骤;如果COOH*的形成是决速步骤,那么便参与了质子偶联电子的过程,因此pH值将对CO2RR产生影响,而CO2*的吸附是决速步骤时,pH将对反应不产生额外的影响;金属Au和Fe-N-C催化剂的限制性步骤是CO2*的吸附,Ni-N-C催化剂和钴酞菁催化剂的限制性步骤是COOH*的形成,通常金属氮碳催化剂具有优异的CO2RR性能主要因为稳固稳定CO2上的大偶极矩47。
3.1.3 无金属碳基催化剂
无金属碳基材料具有独特的固有特性,包括丰富的自然资源、特定的结构、耐高温性质、高比表面积、优良的导电性、环境友好性和对酸碱的惰性。然而,原始的无缺陷碳材料对CO2RR的固有活性可以忽略不计,因为中性碳原子没有能力激活CO2分子。在碳晶格中引入缺陷、空位或杂原子可以改变相邻碳原子的电荷密度和电子结构,有利于优化碳材料中载流子的浓度,提供丰富的活性位点,另外碳有多种同素异构体,可形成不同的杂化形式(sp1、sp2和sp3)48。常见的无金属碳基材料有碳纳米纤维、碳纳米管、石墨烯、金刚石、纳米带、多孔碳等,通常只有掺杂一些杂原子才可用于CO2RR,杂原子类型一般为N、S、B、F、P等49,具有给电子或吸电子特性的杂原子加入到碳中可以显著地调节不同碳的电子性质,从而影响其对CO2RR的催化活性。N原子的尺寸与C原子相似且电负性比碳原子强,因此N原子在碳材料中的掺杂最常见,N掺杂通常有三种形态:吡啶N、吡咯N和石墨N,其中sp2石墨-N是直接取代碳六元环上的某一个碳原子,sp2杂化吡啶-N是连接两个边缘相邻的C原子,并贡献一个p轨道电子形成π系统,吡咯-N在五元环中是以sp3杂化,为π系统贡献了两个p电子,通常吡啶-N被认为是最具催化活性的位点。如将N原子引入碳纳米管(CNT)中,N掺杂CNT不仅具有良好的导电性,还具有大量的吡啶-N缺陷,这些缺陷作为CO2RR的活性位点可降低CO2到CO2*的势垒且增加H+到H*的势垒,N掺杂CNT相比纯CNT,CO2*到COOH*中间体的自由能有了大幅降低,且N掺杂CNT对COOH*中间体有很强的结合能力,而对CO却有很弱的结合能,这主要因为吡啶-N对COOH*的吸附能相比于吡咯-N和石墨-N更负,因此吡啶-N对COOH*中间体的更易紧密结合,而三者对CO*的吸附能比较接近,当过电势为0.18 V时,N掺杂CNT的CO选择性可到达80%50。N原子引入多孔碳纳米片中也可得到类似的结果,N掺杂入碳纳米片中后,与N相连的C将会变得活泼,因此与COOH*中间体结合将得到增强,因此吡啶-N位点可协助碳纳米片吸附COOH*中间体,当过电势为0.49 V时,CO的法拉第效率为84%51。N掺杂的碳纳米纤维作为催化电极和离子液体(EMIM-BF4)作为电解液也可表现出优异的CO2RR到CO性能,这主要因为碳纳米纤维上有两种活性物质:吡啶-N和带正电的碳原子。吡啶-N可与CO2分子弱结合,带正电的碳原子由于具有较高的原子电荷和自旋密度,可作为催化剂将CO2转化为CO,一开始这些带正电的碳原子将被还原,EMIM-CO2将吸附到被还原的碳原子上,碳原子将再次被氧化到起始状态,然后CO脱离这些带正电的碳原子。针对N掺杂的碳纳米纤维,真实的活性位点应是带正电的碳原子,因此在-0.573 Vvs.SHE时,N掺杂碳纳米纤维将CO2还原为CO的电流密度是Ag的13倍52。
3.2 产甲酸或甲酸盐的催化剂
在CO2RR还原的众多产物中,甲酸作为制药和化工原料因其方便的工业储存和运输而被认为具有巨大的竞争优势,其可作为理想的氢载体和液体燃料,也可直接应用于甲酸燃料电池,此外C2以上的产物由于涉及多电子和多质子偶联的反应,这也使得CO和甲酸成为最具有商业化的产物,其中Sn基和Bi基催化剂被公认为最优的产甲酸催化剂。
3.2.1 Sn基催化剂
依据文献报道,催化剂的形貌、尺寸、组分、结晶取向和表面的缺陷都会影响CO2RR的性能。将Sn电沉积到泡沫铜上得到一体化电极,进行CO2RR测试发现产甲酸的法拉第效率和产率相比于纯泡沫铜和纯Sn片都有明显的提高53。先将Sn和Al做成合金Sn20Al80,再将Sn20Al80去合金化后(Al是两性金属可被强碱溶解),得到纳米多孔结构的Sn/SnO2复合物,可表现出高密度的晶界和大量的缺陷位点,以及金属和金属氧化物界面。在所有外加电势下,甲酸的FE都可超过70%,且在-0.8 Vvs.RHE可连续工作58 h54。水热法合成的SnO2微米球也可实现大于62%的法拉第效率55。中空的SnO2纳米球作为催化剂可在500 mA∙cm-2时FE到达75% ±6%,且原位X-ray diffraction (XRD)证实了在催化过程中会被SnO2相还原成Sn单质相56。Wang等57采用液相搅拌法制备得到Sn有机框架催化剂,发现Sn原子与羧基上的O成键,可作为CO2还原的活性位点,根据DFT计算可知,CO2首先会与Sn原子吸附,然后得到一个H生成*OCHO。
除了从催化剂的本身思考外,催化层的厚度和粘结剂(Nafion)的用量也会对CO2RR的性能产生影响。Wu等58改变Sn催化剂的载量来实现催化剂厚度的调控,发现当载量超过4.00 mg∙cm-2时电流密度开始降低,这主要因为此时的催化剂层厚度为9.2 μm,催化剂层超过9.2 μm后活性物质的扩散将受到限制,此外电极中Nafion质量分数为17%–20%时效果最好。Zou等59报道的SnS纳米片也可在120 mA∙cm-2时FE到达88% ± 2%。与SnO2相似,SnS相在CO2RR过程中也会被还原成Sn单质相,但在还原成单质相的过程中,甲酸的分部电流密度和选择性都保持不变,说明了催化剂的活性位点不随形貌和晶相的改变而改变,通过在空气中煅烧可将SnS转变为SnOx,通过二者的CO2RR可发现,S原子的存在有利于抑制氢气反应。Li等60采用水热法合成了珊瑚状SnOx催化剂,发现SnOx的成分是由SnO和SnO2组成,且SnO被厚度为1–2 nm的SnO2所包覆,疏松的SnO2层相比SnO可为CO2还原提供更多的活性位点,此外SnOx相比于纯SnO2样品具有更加优异的CO2还原活性,因此可推测在CO2还原过程中,SnO也起到了正向作用。为了更加明晰CO2还原过程中的活性位点,Zhang等61将SnOx负载到含不同官能团的碳纳米管上。发现当SnOx负载到含羧基(COOH)的碳纳米管上时,SnOx@MWCNTs@COOH电极主要将CO2还原为甲酸,法拉第效率可达到77%;当SnOx负载到含氨基(NH2)的碳纳米管上时,SnOx@MWCNTs@NH2主要将CO2还原为CO。由此可知载体与SnOx之间的界面对CO2还原产物具有调控作用,根据理论计算发现,SnOx与NH2会形成金属-氮碳键,从而有利于吸附*COOH;而SnOx中的O会与COOH中的H形成共价键,从而有利于吸附*OHCO中间体。Liu等62采用SnO2作为催化剂研究了催化剂表面的微环境对CO2RR性能的影响,当催化层中的聚四氟乙烯(PTFE)颗粒过多时,会阻碍H2O扩散到催化剂上,因此CO2还原性能下降,而Nafion存在于催化层中,会改变电子、离子和电子传输通道,故SnO2/PTFE/Nafion在流动式反应器中的电流密度可达到380 mA∙cm-2,甲酸法拉第效率为88.4%,因此可通过在催化层中添加不同的离子聚合物,调控SnO2表面的微环境。催化层中的离子聚合物,一方面可以粘结催化剂,防止CO2还原过程中催化剂脱落,另一方面可作为离子导体,管理催化剂表面的水环境和调控离子浓度。需要注意的是,在长时间电解的过程中,离子聚合物可能会出现溶解,导致催化剂的失活,离子聚合物过多,会降低CO2气体的扩散,还会导致催化剂表面聚集离子,从而抑制CO2RR,因此离子聚合物的选择对于CO2RR的稳定性非常关键。
尽管Sn及ⅥA族元素的衍生物表现出CO2RR到甲酸的性能,但往往FE不超过90%,Sn也可和其他金属复合形成双元或三元金属催化剂,利用双金属的协同作用来提升甲酸的FE。Ye等63合成了异质结构的Sn-Cu合金/Sn,其中SnOx为壳,Sn-Cu为核,在CO2RR的过程中,由于核与壳的相互作用,SnOx仅仅会少量地被还原到单质Sn,因此会原位形成Sn和SnOx的界面,由于HCOO*的形成是放热过程,而*COOH的形成则是吸热过程,因此生成甲酸的决速步骤是HCOO*的加氢形成HCOOH,而生成CO的决速步骤是*COOH的形成,Sn/SnOx界面结合HCOO*和*COOH的能力相比于Sn(211)和SnO2(110)较弱,因此该界面更易往生长甲酸的步骤进行。利用相同的工作原理,Ag-Sn作为核,SnOx作为壳所组成的核壳结构,在-0.8 Vvs.RHE的电势下,SnOx仅会被还原成Sn2+,虽然根据甫尔拜图(Pourbaix diagram),Sn是热力学最稳定的状态,这表明质子还原时嵌入羟基―(O)H*,导致了SnOx层中存在氧空位(oxygen vacancy),CO2在氧空位上的吸附是热中性的,且观察到一个显著的电子转移,表明吸附的CO2是以负离子的形式,即氧空位可稳定OCHO*中间体64。Hou等65利用分步电沉积法制备了Cu@Sn电极并研究了形貌对CO2还原的影响。首先在铜基底上沉积出三维多孔铜基底,然后在多孔铜基底上沉积9层Sn原子,所组成的核壳结构经理论计算,发现可为CO2还原提供更多的活性表面,在-1.33 Vvs.RHE时电流密度可达到55 mA∙cm-2,在-0.93 Vvs.RHE时甲酸的法拉第效率可达到100%,且可稳定工作15 h,说明双金属间的强相互作用非常有利于CO2的活化,并获得优异的产甲酸性能。将Sn和Ga以摩尔比(0.916/0.084)固相混合,基于Sn-Ga的熔点仅为20.4 °C,将Sn-Ga溶液滴在铜基底上可直接作为催化电极。改变电解液的温度为8、13、18和23 °C,电极将实现从固态到液态的转变。发现固态的Sn-Ga电极,甲酸FE不超过40%,但液相的Sn-Ga电极,甲酸FE (23 °C)在-1.16 Vvs.RHE可达到99%。主要原因是Sn在液态的Sn-Ga中以单原子的形式存在,因此活性位点与*OCHO的结合相比于固相得到了增强,因此表现出优异的催化性能66。理论计算表明在Cu(211)面上(图7),甲酸产生的路径为*COOH,但*COOH路径很易进行分离产生*CO + OH*,但*COOH生成反应的自由能高于*COOH解离反应的自由能,但Sn(211)面甲酸的形成遵循HCOO*路径,且该面吸附H*的能力比较弱,因此Sn与*COOH结合较弱,而更有利于产生HCOO*。当Cu(211)边上的3个Cu原子被3个Sn原子取代后,HCOO*加氢的自由能也降低了,表明3SnCu(211)具有较高的甲酸选择性。此外H2在3SnCu(211)面上的形成能得到了显著的提升,故Sn-Cu合金可将CO2还原成甲酸67。Pd和Sn也以合金的形式形成PdSnOx,其中PdSnO2面相比于Pd3O、Pd2SnO和Sn2O2面更有利于甲酸以HCOO*路径生成,因此PdSnO2面对甲酸的选择性最高68。
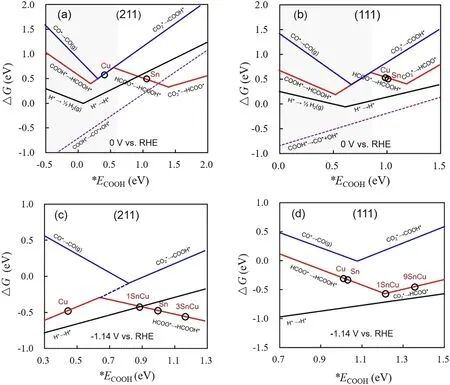
图7 在不同电势下过渡金属表面上*ECOOH的自由反应能67Fig.7 Calculated free reaction energies as a function of *ECOOH on transition metals at different potentials 67.
3.2.2 Bi基催化剂
具有低维活性位点的Bi枝晶可使CO2还原到甲酸沿着*OCHO路径。理论计算表明高指数晶面(012)、(110)和(104)相比于(003)面可更好地稳定*OCHO中间体,其他高指数晶面如(107)、(116)、(214)和(009)也对CO2的还原有正向作用69。Bi枝晶也可产生晶界,利用晶界间的应力促进CO2RR,Chen等70利用溶解在电镀液中的Zn2+合成富含大量晶面的Bi枝晶,通过同步辐射发现Bi―Bi之间的键长为2.85 Å (1 Å = 0.1 nm),小于Bi片3.03 Å,证实了晶体之间存在应力效应,原位电化学衰减全反射傅里叶变换红外证实了富含晶界的Bi枝晶有利于稳定*OCHO中间体。但Bi纳米片对于CO2RR到甲酸的机理则和Bi枝晶完全相反,Zhang等71合成了厚度为1.2–1.5 nm的超薄纳米Bi片,并发现该Bi纳米片主要暴露(003)面,由于Bi纳米片具有大量的边活性位点可用于催化CO2,这些活性位点可促进*OCHO的形成,且Bi(003)面将CO2转化为*OCHO的自由能为-0.41 eV,而(012)面的自由能高于Bi(003)面,说明了(003)面在动力学上更有利于CO2RR。将超薄BiOI纳米片(厚8.7 nm)电化学原位还原为Bi超薄纳米片,通过高分辨透射电子显微镜可知,Bi纳米片表面暴露了大量的(110)晶面,在Bi纳米片上,*OCHO形成过程的自由能是+0.49 eV,COOH*和H*的自由能分别是+1.16和+0.95 eV,因此Bi纳米片更有利于甲酸的生成且甲酸的FE可接近100%72。BiCuSeO超晶格纳米片中的Bi是以三价的形式存在,即使在CO2RR过程中Bi仍是以氧化态的形式存在,但在BiCuSeO中Bi―O的键长大于其在Bi2O3中的,因此Bi―O在BiCuSeO具有独特的结构,理论计算表明该特殊结构的Bi―O有利于吸附*OH和*OCHO中间体,而亚层的[Cu2Se2]2-非常有利于电子的转移,因此BiCuSeO超晶格纳米片甲酸的FE可达90%73。Guan等74采用水热法将In掺杂入Bi2O3中,DFT理论计算发现In的引入可降低OCHO*的形成能,因此相比于纯Bi2O3更有利于将CO2还原成甲酸。Zhang等75用溶剂热还原法合成了微米结构的Bi催化剂。相比于商业的Bi基催化剂,所制备的Bi催化剂对CO2RR具有更好的活性,甲酸法拉第效率达90%,过电势仅为600 mV。这说明尽管Bi催化剂的晶体结构没有改变,形貌的不同也会影响CO2RR的性能。基于此,Zhang等76进一步用乙二醇作为稳定剂,成功将微米结构的Bi催化剂调控为纳米尺度(50–100 nm),表现出更加优异的CO2RR性能,在-0.83 Vvs.RHE时,甲酸的法拉第效率提高到了94.7%,且可稳定工作20 h性能几乎无衰减;另外,还研究了Bi基催化剂的失活机制,发现金属态的Bi在进行CO2还原的过程中,会逐渐转化成(BiO)2CO3,从而导致CO2RR活性的降低,因此该失活机制对后续Bi基催化剂的设计具有重要的指导意义。
3.2.3 In基催化剂
Huang等77研究了纳米In2O3立方体的尺寸效应,发现15 nm的In2O3立方体甲酸法拉第效应可达95%,而当尺寸降低到5 nm时,还原反应主要是氢气的析出。Pan等78将纳米In2O3颗粒利用水热法负载于碳纤维上,发现在CO2电催化还原的过程中,In2O3会被还原成In金属,在H槽式和流动式反应器中,甲酸的法拉第效应都可达到90%以上。Du等79利用磁控溅射技术合成了带氧缺陷的In2O3薄膜,发现氧缺陷的存在可提高材料表面的应力应变,且氧缺陷与In原子的电荷转移也可得到增强,缺陷的数量越多电荷转移的强度越大,因此较强的电荷转移强度有利于CO2的活化和*OCOH的吸附,故甲酸的法拉第效率在-0.85 Vvs.RHE时可达70%以上。除了In2O3可将CO2高效还原为甲酸外,Grigioni等80合成了InP量子点并将其负载于疏水的碳布上,由于InP量子点经6-巯基己-1-醇配位处理,所以在CO2还原的过程中,InP会被转变为S保护的In2O3和In金属单质,表面的In金属是CO2还原的活性位点,S位点的存在有利于水的电解并为CO2还原提供质子,In金属和S位点的接触处也有利用吸附H*和*OCHO中间体,因此该电极在三腔室反应器中产甲酸盐的分电流密度可达930 mA∙cm-2,同时甲酸的法拉第效率为90%。基于S位点有利于水的电离,但在电催化CO2的过程中,S会从In2S3中析出到电解液中,导致催化CO2的性能衰减。Chi等81将Zn引入In2S3纳米花中,发现Zn的存在可改变In2S3的配位环境,从而调控In2S3的电子结构和催化性能,In2S3存在大量的四面体空位,而Zn的引入正好可以占据这些空位,导致Zn原子通过四面体配位与S原子结合,四面体结构通常具有共价特征,这意味着Zn―S键得到了增强,因此Zn的引入导致了ZnIn2S4具有非常稳定的结构,同时In与S之间的电子传递也因Zn的引入得到了增强,故ZnIn2S4作为催化剂在-1.18 Vvs.RHE时,将CO2还原为甲酸盐的法拉第效率达到99.3%,电流密度在流动式反应器中获得298 mA∙cm-2且稳定性可维持在60 h。
4 电解液对CO2RR性能的影响
除了催化剂作为影响CO2RR性能的主要参数之外,电解液的种类也会对CO2RR产生巨大影响82,其主要原因是电解液中阴离子和阳离子的不同浓度会引起不同的静电相互作用、缓冲容量、pH值和质子供体,这些因素往往相互作用,很难将具体某一因素的影响详细探讨,以下将简要介绍pH、阳离子和阴离子对CO2RR的影响。
4.1 pH值的影响
CO2作为酸性分子溶于电解液后会形成碳酸氢盐和碳酸盐,因此CO2RR通常是在中性或酸性条件下进行,1989年Hori等83首次通过实验揭示了CH4和C2H4的选择性之间受pH影响。Wang等84研究了pH为7和13时CO2RR性能的差异,在标准电极电势下(SHE)研究了产物选择性和过电势的关系,发现C1产物(如CH4)的过电势表现出与pH的相关性,因为甲烷的形成涉及多步质子偶联电子的过程,而C2+产物的限制性步骤与pH无关,可能的因素有相电子转移的速度、质子转移的速度、反应过程中间体形成的过程(C=C偶联反应)、从水中得到质子和电子的能力、在溶液中电子的转移速度远大于质子的转移速度,因此该部分不是限制性步骤,从水中得到质子和电子的能力与pH值相关,C2+的限制性步骤是C=C偶联反应,因为此过程没有质子的参与85。对于CO2RR最佳性能来说pH值应该有适当的区间,相关文献报道认为最佳微环境的pH值应该是9–10,因为在高pH值下CO2会形成碳酸氢盐和碳酸盐导致微环境下的CO2反应态较少,此时的HER将成为主导,如果反应不受CO2传质的影响,如C2+的形成过程,则pH的影响并不大86。而在酸性条件下,CO2将不会和电解质形成碳酸氢盐和碳酸盐,Huang等87用阳离子增强CO2活性策略研究了CO2RR在强酸条件下的产物分布,发现在电流密度为100 mA∙cm-2和pH值为1时,往电解液(1 mol∙L-1H3PO4)中加入0.5 mol∙L-1KCl时,HER和甲烷的选择性都有所下降,当加入不同浓度的KCl时,发现塔菲尔斜率有所变化,这说明pH和CO2RR反应独立,也证明了在铜电极上CO2RR的速率决定性步骤是CO2的活化,当电流密度小于200 mA∙cm-2时,K+对于CO2的活化和HER的抑制几乎不起作用,但在400 mA∙cm-2时,反应将从HER向CO2RR反应转变,这说明在强酸和大于200 mA∙cm-2条件下,阳离子可诱发CO2的活化。Fu等88以SnO2纳米球为催化剂研究了pH值对CO2RR性能的影响,发现当电解液的pH值为6时,阴极侧发生的反应主要是氢气的析出,而当电解液的pH值为9时,高浓度的碱性环境也不利于CO2还原至甲酸,当电解液的pH为8.3时,电化学还原CO2将具有最大的电流密度,这说明CO2更易在弱碱性环境下被还原,且在弱碱性条件下,SnO2也可保持很好的稳定性。
4.2 阳离子的影响
Hori和Murata首先证实了CO2RR的活性和选择性受电解液中碱金属阳离子的影响,即CO2在多晶铜上的选择性受阳离子大小的影响,较大的阳离子会增加C2+的选择性并降低HER的选择性,在0.1 mol∙L-1MHCO3(M = Li+、Na+、K+、Cs+)水溶液中,HER的活性趋势为Li+> Na+> Cs+> K+,而C2+的活性趋势与HER不同,为Li+< Na+< K+ 目前CO2RR领域应用最广泛的是KHCO3电解液,CO2可与碳酸盐-水保持平衡,有助于维持中性环境,因此,阴离子的效应往往忽略。Hori等83的研究工作发现CO和乙醇更易在KCl、KClO4、K2SO4和稀KHCO3中产生,而在K2HPO4中主要进行HER反应。在之前讨论过的在CO2RR过程中,在电极附近会产生大量的OH-,而HCO3-和H2PO4-可以和OH-进行中和反应。相反KCl、KClO4和K2SO4由于没有中和物种,溶液的pH值将得到升高,故有利于CO2RR反应抑制HER,同时有利于C2+反应而不是C1反应。我们的研究工作也证实了HCO3-浓度对CO2RR产甲酸性能的影响,即甲酸的产率和法拉第效率都随着HCO3-浓度的增加呈现出先增加后减小的趋势,而活性则一直增加92。Zhu等93用衰减全反射表面增强红外吸收光谱学研究了HCO3-在CO2RR过程中的关键作用,证实了碳酸氢根不仅仅是简单的pH缓冲液或质子来源,而是参与到了CO2到HCO3-的过程中,即CO2必须先溶于溶液中形成HCO3-实现平衡后,才可参与到CO2RR。事实上,产物选择性除了受到电解质阴离子的影响外,催化剂中的阴离子同样会对CO2RR过程有重要影响。Xu等94通过阴离子工程调控实现Cu/In/MOF双金属高效电催化还原CO2,发现SO42-阴离子前驱体更有利于甲酸产物的生成,而NO3-阴离子更有利于CO产物的生成。在-0.86 Vvs.RHE电位下,0.5 mol∙L-1KHCO3电解液中Cu1In3-MOF-NO3具有较高的CO法拉第效率,而Cu1In3-MOF-SO4在-1.16 Vvs.RHE时则表现出最优的甲酸盐法拉第效率。 如图8a所示,H槽式反应器由于易于安装被广泛用于筛选催化剂95。H槽式反应器中的“H”起源于装置的形状,它包含阴极腔室和阳极腔室,两个腔室通过隔膜隔开,通常在H槽中采用三电极测试,即工作电极和参比电极在阴极腔室,对电极在阳极腔室,工作电极即制备的催化剂涂敷于导电基底上,参比电极类型应用最广泛的是银/氯化银、汞|氧化汞电极、甘汞电极等,参比电极的选择根据阴极电解液的pH值而定,对电极常使用铂丝、铂片、碳棒、泡沫镍等。H槽式反应器缺点是电极面积往往不超过9 cm2,且电解液在腔室中更换很不方便,由于受腔室的空间所限,CO2RR进行过程中易受传质的限制,因此,H槽式反应器中的电流密度往往不超过100 mA∙cm-2。虽然在阴极腔室加入转子将阴极电解液搅拌可在一定程度上改善传质效应,但仍很难实现大于100 mA∙cm-2的电流密度。 图8 (a)传统H槽示意图;(b) H型的流动式反应器示意图;(c)微流控反应器示意图;(d)气腔室反应器示意图;(e)膜基反应器示意图;(f)三腔室反应器示意图Fig.8 A schematic depicting selected (a) conventional H cell; (b) H-type flow cell; (c) microfluidic reactor Zero-gap cell;(d) gas-chamber reactor; (e) zero-gap cell; (f) three-compartment reactor. 如图8b所示,具有连续电解质的H型反应器和传统H槽式反应器具有相同的组件,不同的是H型流动式反应器是将阴极腔室和阳极腔室的电解液在外接泵的基础上循环流动起来,这样做的优势在于可以增加阴极和阳极的传质,更换电解液也更便利。如Kuhl等96用自制的H型流动式反应器得到了多达16种CO2还原后的产物,为了实现均衡的电场分布,在该装置中阴极电极和阳极电极被平行放置,且阴极电极的面积小于阳极电极的面积,工作电极的面积为1.5 cm × 3 cm,容量为8 mL,CO2的流速是20 mL∙min-1,选择20 mL∙min-1的流速是为了确保将CO2充分输送至电极表面,同时防止气泡撞击电极表面造成干扰。值得一提的是,在该装置中阴离子交换膜被用于阻隔阴阳两腔室,但该膜不能完全阻止甲酸和乙酸从阳极到阴极的穿透损失,CO2进入反应器之前通过水进行加湿可减少挥发性液相产品的蒸发,Ag/AgCl用作参比电极,参比电极与工作电极的距离仅仅为0.5 cm,电解液为0.1 mol∙L-1KHCO3。Peng等97对比了H型反应器与流动式反应器的性能差异,阴极电极为生长于氮掺杂碳布上的CuSn合金,CuSn合金电极的有效面积为2 cm2,阳极电极为铂片,铂片的面积大于CuSn合金电极,阴极电解液为CO2饱和的0.5 mol∙L-1KHCO3,阳极为0.5 mol∙L-1KOH,且阴阳两极的电解液在蠕动泵的作用下以13 mL∙min-1的流速循环流动,CO2还原的气体和液体产物由KHCO3的流动从阴极室引出并检测含量,发现流动式反应器的电流密度为66.41 mA∙cm-2,相比于H型反应器的15.56 mA∙cm-2提高了4.2倍,并可稳定工作20 h,甲酸产率可达到163 μmol∙h-1∙cm-2,且槽压仅为3.17 V。 微流控流动式反应器是由Kenis于2010年提出的98。如图8c所示,该结构的特点在于阴极和阳极之前没有隔膜,仅依赖于一个非常薄(< 1 mm)的流体通道,将阳极和阴极的液体电解质隔开。CO2气体直接扩散到催化剂上,催化产物在气体扩散电极和电解质之间产生,这种结构的阳极侧和阴极侧无需膜进行分隔,而是依靠气体产物的扩散来分离还原产物和氧化产物,通过精确控制微流控反应器的各种参数,可达到CO2RR的高电流密度。这些参数包括:电极层的沉积方法,扩散层的组成,电解液pH值和组成成分等。2012年,Kenis等99用氨配体Ag/C作为催化剂,催化剂载量为1 mg∙cm-2,中间流体为1 mol∙L-1KOH,CO的分部电流密度在-1.8 Vvs.Ag/AgCl时可达95 mA∙cm-2。随后,该团队进一步研究了不同电极的制备方法对CO2RR性能的影响,尽管手喷和空气喷法制备的气体扩散电极形貌差异很大,但总电流密度没有明显的差别。原因可能是微流控反应器独特的结构,使电解液可有效地进入了催化层中,其详细结构是阴极和阳极气体扩散电极对面放置,中间流体层的厚度为0.15 cm,宽0.5 cm,长2.0 cm,电极面积为1 cm2,集流体是两个铝片,CO2气体直接通到催化电极的背面,阳极背面与空气连通,装置由4个螺栓固定,CO2的流速为7 mL∙min-1,中间层电解液(1 mol∙L-1KCl)的流速为0.5 mL∙min-1100。Dinh等人101在Kenis团队的基础上进行了改进,采用石墨纳米颗粒组成的阴极扩散电极结构,并在其表面引入一层PTFE,中间层是100 nm厚的Cu催化层,与电解液接触的是PTFE层,这样的结构可以确保CO2RR在强碱条件下进行,即中间溶液为10 mol∙L-1KOH,PTFE与电解液间的突出界面以及高浓度的KOH可实现CO2RR在特定区域发生,且通过实验发现Cu催化层厚度越薄越有利于CO2RR产乙烯。有趣的是尽管加入石墨纳米颗粒作为集流体对整体装置的电流密度没有提高,但乙烯的选择性却提高到70%,这主要因为石墨层改善了CO2气流的分布,更有利于CO2RR的进行101。 如图8d所示,气腔室反应器是在微流控反应器的基础上在中间层溶液中引入一层隔膜。隔膜的引入可使阴极电解液和阳极电解液保持不同,且阴极腔室产生的液体产物不会扩散到阳极被氧化,阴极气体扩散电极一侧与电解液直接接触,背面通入CO2气体,阳极气体扩散电极一侧与阳极电解液直接接触,背面与空气连通。这种结构可以克服很多CO2RR面临的缺点,如在高电流密度下CO2密度较低、H+从阳极到阴极的穿透或OH-从阴极到阳极的穿透带来阴极电解液pH值的变化。此外,可以很方便地将参比电极放入阴极电解液中,防止阴极产物向阳极的扩散以及实现阴极气液两相产物的同时检测等。但仍需要注意的是,这种类型的结构也存在很多挑战,如阴极气体扩散电极的疏水性在长时间运行过程中容易失活,造成水淹现象;该结构的维持和CO2气腔室的压力有很强的相关性,如较大的气压将使催化层和电解液界面断层,造成短路现象。另外由于阳极侧与空气直接相通,较大的CO2气压也会使阳极侧的反应受到干扰;但较小的CO2气压又不能供应足够的新鲜CO2参与还原反应102。Lu等103利用气腔室反应器在113 mA∙cm-2时获得82%的甲酸法拉第效率,反应器阴极和阳极容量均为0.6 cm × 0.9 cm × 0.3 mm,反应的有效面积为0.2 cm × 0.5 cm。 在气腔室反应器中催化电极的结构会直接影响CO2RR的性能,该催化电极也被称为气体扩散电极(GDL),通常是将催化剂直接负载于疏水多孔的导电基底上,CO2扩散到催化剂表面的长度仅为50 nm,相比于H槽的50 μm,GDL催化剂表面可以维持很高的CO2浓度,因此可在大电流密度下,快速催化CO2,但CO2还原必须在水参与下完成,故合理设计三相界面处催化剂结构至关重要104。气腔室反应器中阴极电解液可为高浓度碱性溶液,碱性电解液可极大地抑制竞争反应HER,但同时未参与还原的CO2会与OH-反应生成CO32-或HCO3-,造成碳损失,因此需要调控催化剂表面的微环境,实现CO2还原高性能的同时,还要避免碳损失。管理催化剂表面的水可实现对微环境的调控,催化剂表面的水含量会影响CO2或产物与电解液的接触,进而影响电化学反应动力学、气体扩散途径和电子传输路径,通常认为疏水的微环境可避免催化剂活性位点与电解液的过多接触,创造出CO2或产物的传输通道,该通道的存在被认为有利于C2+产物的生成105。如Xing等将商业铜颗粒与PTFE做成催化层,通过改变PTFE的含量,C2产物的分部电流密度可达到> 250 mA∙cm-2,最大转化率为14%,是未加PTFE的2倍,该工作证实了疏水的微环境可以提高CO2到C2+产物的还原效率106。除了管理催化剂表面的水,具有分级多孔的催化层也可使CO2RR保持在较高的转化率,三维的孔结构不仅可以提供更多的活性位点,反应物和产物也可在孔结构中扩散,电子和离子也可更好地在孔结构中传导107。但应注意的是,尽管越厚的催化层会暴露越多的活性位点,但质子和CO2在催化层中的扩散也会受到影响,从而降低CO2的转化率108。此外,还可在催化剂层与基底之间,引入一层微米孔疏水层,该层的存在可允许气体分子通过,而限制电解液的渗透,既避免了水淹现象的产生,也可降低GDL的电阻109。导电基底的厚度也会影响CO2RR的活性,如Ma等110采用Ag基GDL研究了碳纤维导电基底的厚度对CO2气体扩散的影响,发现当基底的厚度降低时,更有利于CO2气体的扩散,但也更容易发生水淹现象,导致催化性能快速下降,因此合理调控导电基底的厚度也至关重要。 如图8e所示,膜基反应器顾名思义阴极和阳极被膜隔开,由于阴极和阳极的距离只有膜的厚度,因此又可称为零距离式”反应器。此外,由于阴极没有使用电解液,故通入的CO2必须要经过加湿处理,阳极的电解液可用于保持膜的湿润。膜基反应器有很多优势,如阴极侧无电解液减少了配件泵的使用、CO2流场更易调控、可对CO2气体加湿和增压、欧姆电阻较小等,因此膜基反应器将是未来产业化CO2RR研究的重点。此外,阴极不使用电解液也排除了阴极气体扩散电极被水淹的可能性,也可避免电解液与催化剂直接反应造成失活,同时可避免碳酸盐的形成,但仍需要注意液体产物的聚集会阻碍气体的扩散,如何尽快排出液体产物仍是一个挑战。 根据膜的类型,膜基反应器可分为阳离子膜基、阴离子膜基和双极膜等三类反应器。目前广泛应用的是阳离子膜基反应器。阳离子膜基反应器的工作原理是H+或其他类型的阳离子在电场力的作用下,从阳极到阴极进行穿透。如Yin等111用疏水碳纸作为阴极集流体,Au/C催化剂直接涂于阳离子交换膜上,阳极则将IrO2催化剂制备在Ti网上,在电流密度为500 mA∙cm-2时,槽压仅为3.0 V,CO的法拉第效率可达到85%。阳离子膜基反应器的最大缺点是由于阳离子强质子传导机制,导致阴极催化剂处于强酸性环境,因此要求催化剂耐强酸以避免失活,同时,长时间的强酸环境也会有助于HER反应。阴离子膜基反应器可避免阳离子膜基反应器的缺点,同样将Au/C催化剂涂于阴离子膜上,当槽压为2–2.4 V时CO的法拉第效率可提高到95%。阴离子膜基反应器最大的缺点是,CO2会与从阳极传导过来的OH-结合形成CO32-,而CO32-则从阴极到阳极的穿透,不仅造成碳损失,还会增加阳极的OER反应动力学。双极膜基反应器可解决阴阳离子膜基反应器的共同缺点。Blommaert等112对比了阴离子膜和双极膜在膜基反应器中的CO2RR性能差异,具体参数为:气体扩散电极的面积为6.25 cm2,有效工作面积为5 cm2,Ag催化剂直接涂于碳纸上,催化层的厚度为100 nm,泡沫镍作为阳极催化电极,阳极电解液为1 mol∙L-1KOH,加湿的CO2流速为40 mL∙min-1。研究发现在阴离子膜基反应器组件中,尽管槽压不高,但CO2到阳极的损失导致阳极pH下降,进而造成整体的能量利用效率下降,在双极膜基反应器中阳极的pH可保持恒定,但双极膜基反应器的总耗能仍高于阴离子膜基反应器。Liu等113采用铜基有机框架作为阴极催化剂,阳极采用负载于疏水碳纸上的IrO2,Nafion 117为中间隔膜,组装入膜基反应器中,发现在-0.64 Vvs.RHE时,电流密度为34.97 mA∙cm-2,是H槽的2.8倍,在-0.87 Vvs.RHE时,电流密度达到230 mA∙cm-2,为膜基反应器应用于CO2RR的商业化提供了可能性。 如图8f所示,三腔室顾名思义应具有三个腔室,分别为阴极腔室、阳极腔室和中间腔室,其中阴极腔室和中间腔室之间由阴离子交换膜隔开,阳极腔室和中间腔室之间由阳离子交换膜隔开,中间腔室通常是由多孔的固态电解质组成,可允许去离子水流收集生成的液体燃料,也可促进离子的传导。固体电解质可以由离子导电聚合物或无机离子导体构成,还可以是导电阳离子或导电阴离子,CO2RR过程中,产生的甲酸根(HCOO-)和乙酸根(CH3COO-)在电场力的作用下可穿过阴离子交换膜到中间室,阳极OER过程中产生的H+也可在电场力的作用下穿过阳离子交换膜到中间腔室。因此,HCOO-或乙酸根CH3COO-会与H+在中间腔室进行离子复合生成HCOOH和CH3COOH。2017年Masel团队首先将该套装置用于生产纯甲酸,阴极催化剂采用Sn负载于50% PTFE疏水的碳纸上,阳极是纳米IrO2负载于5% PTFE疏水的碳纸,阴离子交换膜为Sustainion系列膜,阳离子交换膜为Nafion系列膜,中间层离子树脂为Amberlite® IR120 (hydrogen form,Sigma-Aldrich);Dowex® 50WX2 (hydrogen form Sigma-Aldrich);Dow Amberlite® IRN-77 (ion exchange resin,nucleargrade,VWR Scientific); Duolite® Resin C433 (Alfa Aesar),该套装置可在140 mA∙cm-2的电流密度下连续工作500 h,且槽压为3.5 V,可得到纯甲酸的质量分数为5%–20%114。Xia等115在Masel团队的基础上,设计了含有不同官能团的固态电解质,如磺酸官能团和季胺官能团组成的多孔苯乙烯-二乙烯基苯共聚物,该固态电解质不仅可传导H+还可以传导HCOO-,阴极采用二维的Bi纳米片作催化剂,阳极采用镍铁LDH做催化剂,整套装置在163 mA∙cm-2时,甲酸法拉第效率可达73.3%,且槽压仅为1.33 V。 在气相CO2流动式反应器中已经有三种膜,分别为:阳离子交换膜(CEM)、阴离子交换膜(AEMs)和双极膜(BPM)。每种类型的膜应用在阳极和阴极之间都有不同的离子传输途径,如图9所示。通常认为官能团的浓度、膜的厚度、含水率和离子电导率会影响膜的性能,因此在选择膜的类型时,需要根据装置两侧的电解液条件(如pH、浓度、离子类型)来决定,以有效地调节水和离子的传输并促进电极表面发生的质子偶联电子转移反应。 图9 三种不同膜的传输方式Fig.9 Overview of the different ion transport pathways. 最常用的阳离子交换膜是质子交换膜(Nafion系列膜),包括如Nafion®115、212、117和324等。2008年,Newman和他的同事报告了一种Nafion膜基CO2流动式反应器,该反应器可将CO2还原为CO,这也是第一个基于多聚物电解质膜的流动式气相CO2电解的装置。为了抑制H2的形成,必须在膜和阴极扩散电极之间加入一层碱性电解质缓冲层(1 mol∙L-1KHCO3),但在100 mA∙cm-2的电流密度时,法拉第效率仅为20%,且稳定性仅能维持几个小时116,这些结果证明了在气相流动式反应器中水和离子传导的重要性,因为膜的性质与含水率和离子含量直接相关。 碱性阴离子交换膜的工作原理是负离子(例如OH-)从阴极到阳极的传导。这种离子传导机制比质子交换膜的传导机制更适合CO2RR,因为在不向阴极输送H+的情况下,可避免阴极的HER反应,从而可促进CO2到产物的正向反应。一般情况下从阳极传导到阴极的OH-可与CO2快速生成HCO3-和CO32-,可直接参与CO2RR反应,需要注意尽管HCO3-和CO32-的离子迁移率比OH-低得多,但如果这些较大离子在膜中的积累过多,也会抑制膜中OH-的迁移,降低CO2还原效率。此外,HCO3-和CO32-从阴极转移到阳极会降低电解槽的整体效率。尽管如此,目前已知的许多性能最好的膜基流动式反应器仍采用碱性离子交换膜。在2017年,Masel等117以N-甲基咪唑取代苯乙烯共聚物作为碱性离子交换膜,在-3.0 V时电流密度可达200 mA∙cm-2,CO的法拉第效率达到了90%且可连续电解4500 h。Peng等118采用Bi-Cu双金属作为阴极电极,石墨棒作为阳极电极,研究了酸性膜(Nafion 117,美国杜邦公司)和碱性膜(FAA-3-PK-130,德国Fumasep公司)对CO2RR性能的影响。发现当Nafion 117作为隔膜时,在-0.91 Vvs.RHE时,产甲酸的最大法拉第效率为89.93%,而当使用FAA-3-PK-130作为隔膜时,甲酸的法拉第效率可以提高到94.37%,因此碱性膜相比于酸性膜更有利于将CO2还原为甲酸。此外,碱性膜中聚合物骨架所含官能团对CO2RR性能同样会产生很重要影响119。Wang等120研究发现,当碱性膜表面带噻吩基团时,相比于带二甲辛基和羟苄基团,更有利于CO2还原为甲酸。这主要是噻吩基团可使碱性膜的表面更加平整和紧密,致使碱性膜具有非常优异的离子电导率和较低的含水率。进一步在碱性膜中引入咪唑环结构,发现所制备的碱性膜经热处理、化学交联和离子交换后呈现三维半互穿多孔网络结构。该网络结构在进行离子交换时可捕获更多的OH,这些捕获的OH通常以氢键的形式或物理吸附在碱性膜的表面,因此可为OH-提供传输通道;此外,咪唑基五元杂环结构的存在也是碱性膜具有优异化学稳定性的原因,而吡咯烷酮基团上的氮原子相比于碳原子具有更高的电负性,可以提高相邻碳原子的电子云密度,促进膜内结构的稳定,该双重效应使制备的碱性膜具有优良的OH-电导率(21.47 mS∙cm-1)、较低的含水率(57%)和较高的离子交换容量(0.91 meq∙g-1);当该碱性膜应用于CO2RR时,相比于Nafion 117和碱性膜(A201,日本株式会社)都具有更好的性能,体现在具有最大的电流密度(55.37 mA∙cm-2),最佳的甲酸法拉第效率(88%)。Wu等121采用分层法和双交联工艺法相结合的方式,以不同分子量的聚二烯丙基二甲基氯化铵(PDDA)和聚乙烯醇(PVA)为改性剂,合成了以细菌纤维素(BC)为基底的碱性膜,PVA的引入使碱性膜内部形成一个稳固的内部三维网络状结构,克服了BC膜松散的问题,在常温常压下,碱性膜的OH-电导率可达到72.95 mS∙cm-1,当该膜用于CO2RR时,在-1.06 Vvs.RHE时,电流密度可达到47.44 mA∙cm-2,甲酸法拉第效率为67.89%,连续工作20 h后,性能仅衰减7.09%。使用碱性膜不可避免地会造成CO2还原产物、HCO3-和CO32-从阴极向阳极传导,改善这种传导效应可通过降低碱性膜的含水率或提高带正电的基团,含水率过高会导致膜的溶胀,产生应力作用在催化剂上,限制CO2与催化剂的直接接触,过低的含水率又会影响OH-的电导率,因此含水率应控制在合理的水平。Wang等122在碱性膜中引入氧化石墨烯来提高OH-的电导率并维持合理的含水率。氧化石墨烯表面含有丰富的含氧基团,可与聚合物基体以氢键的方式结合,形成连续的长距离OH-迁移通道,亲水的石墨烯也可维持碱性膜的含水率。因此引入氧化石墨烯使CO2RR电流密度在-1.16 Vvs.RHE时提高到43.5 mA∙cm-2,甲酸的法拉第效率达到90%,远高于A201的80.2%和Nafion 117的58.4%。 双极膜的组成包括带正电荷的阴离子交换层(AEL)、中间催化层和带负电荷的阳离子交换层(CEL)123,它可以在两种模式下工作:(1)正向偏压(V> 0),此时膜的CEL朝向阳极;(2)反向偏压(V<0),此时CEL朝向阴极。在正向偏压模式下,电场导致可移动离子向界面区(IR)迁移,离子在结处聚集从而补偿层中的电荷,进而降低膜的选择性。相反,在反向偏压的模式下,当施加的外加电压到达某一值时,在AEL和CEL的界面处将发生水分子的分离,且在膜的内部存在一个增强电场效应,即第二维恩效应的Onsager定律124,在外加电场的存在下,H+将通过CEL向阴极迁移,OH-通过AEL向阳极迁移。相比单层碱性膜或酸性膜,双极膜在电化学还原CO2中具有突出优势:(1)双极膜作为隔膜时,阴阳两极可以使用两个不同pH值的电解质溶液,且在使用过程中可以维持两侧电解质的pH;(2)液体产物从阴极到阳极的“穿透”(crossover)可以忽略;(3)当阴、阳两极的电解质为纯水时,电解质的酸化和碱化无需添加额外的酸和碱。目前双极膜也被用于CO2RR,如2021年Mallouk等125研究了双极膜在零距离反应器中对气体产物CO的影响,发现膜与电解液接触的微环境对CO的产生有重要的影响。Smith等126利用银基催化剂和中性电解质,发现使用双极膜作为中间隔离层,整体槽压相比于Nafion膜可降低1 V。Peng等127采用锡掺杂铋作为阴极电极,泡沫镍负载的铁作为阳极电极,研究了商业碱性膜和自制双极膜对CO2RR性能的差异。发现当使用碱性膜作为隔膜时,阴极CO2还原的甲酸盐会穿透碱性膜到阳极,穿透率为8.9%,而当使用自制的双极膜时,在阳极检测不到甲酸盐的存在,说明甲酸的穿透得到了很好地抑制,且在双极膜的阴极侧观察到大量的再生CO2气体。CO2气体的出现是中间缓冲层中水解离产生的H+穿透了阳离子交换层,达到了膜与阴极电解液的界面,产生了一个酸性环境,该界面处的高浓度H+会与碳酸氢盐反应,形成CO2气泡。尽管如此,双极膜两侧通常具有较大的电势差,因此在反向偏压下电解时需要更大的电压驱动反应,造成整个体系内阻的升高,从而能源利用效率较低。 在我国宏观政策的刺激下,CO2资源化领域必将成为未来科学研究重点。尽管电催化CO2的相关研究起步于上世纪80年代,但该领域前三十年的发展非常缓慢,近十年来无论是从理论还是实验方面,都取得了长足进展,也引发了广大科研工作者的兴趣。但如何将该技术落实并造福人类需要科学界和工业界的共同努力,未来电催化CO2还原的产业化和工业化需要从以下方面进行开展: (1)高性能催化剂的开发。设计的催化剂需要在高产率的情况下具有较高的能量密度,且针对不同的还原产物,催化剂的表面对不同中间体的吸附能也要有所差异,若是C2+以上的产物,催化剂需要对*CO中间体的吸附能较强,才可进行后续C=C偶联反应,而实现*CO中间体的吸附可对催化剂的形貌和电子结构进行设计,如催化剂可暴露出更多的低维活性位点,通过调控催化剂的孔结构,实现*CO中间体的限域,掺杂或合金化Cu基催化剂,改变局域电子结构等。从经济的角度思考,应用到工业界的催化剂,特定产物的法拉第效率需要超过90%,槽压低于1.8 V,且电流密度要超过300 mA·cm-2,稳定性要超过8000 h,有望实现这些指标的方法是设计独特形貌的催化剂、合金化多种金属、诱导表面的晶格应变、调控催化活性位点的电子结构及功能化催化微环境等。此外,催化剂的制备方法也要环保且耗时短,并可批量化和规模化生产。 (2)深入研究反应机理。对CO2RR的反应机制也应理论化,从而摆脱目前试错式催化剂的制备,应实现用理论指导催化材料的合成,从而快速合理地制备高活性催化剂。第一性原理计算(DFT),通过改变模型中的原子结构、电解液性质和电场分布等实际参数可更深入和可靠地理解反应机理。此外,高通量的机器学习也有利于精确地建立DFT模型,从而更进一步地引导计算和过程优化。由于催化剂在CO2RR过程中也会发生物相转变或者与电解液发生反应,导致按照原来的晶体结构计算的结果与实际并不相符,因此未来需要采用高通量计算并与机器学习进行结合,更加精确地模拟催化剂表面的动态过程。 (3)电化学反应器的开发。CO2RR工业化至关重要的一步是设计大面积反应器,或者将若干个反应器串联和并联成反应堆。无论是大面积反应装置还是反应堆,最核心的问题是反应装置的放热,导致装置的能量利用效率低,且温度的升高也会诱导催化剂的失活,因此需要解决反应装置的放热问题。CO2为气体,无论是转化为液体产物还是气体产物都要经历若干个过程,在转化过程中往往有相变的发生。CO2气体需要先溶于电解液中且要快速补充到活性位点上,而还原产物则要快速扩散远离活性位点,因此需要设计更加合理的流场分布和电场分布。 (4)目前常用的电解质溶液仍以碳酸盐类为主,开发新型的电解质溶液是非常有必要的,且该种电解质还应满足以下几点:(i)需大量溶解CO2,从而有利于CO2更多地参与反应被还原;(ii)抑制CO2还原反应的竞争机制析氢反应;(iii)电解质需对环境友好,且使用过程中具有稳定的化学性质,无安全隐患。 (5)作为CO2电化学还原的核心部件,膜的作用不言而喻,开发高性能的膜可阻止还原产物的透过损失,且膜的耐用性也非常重要。针对阳离子交换膜将阳离子从阳极传导到阴极的机制,会导致阴极侧pH值降低,故会有利于氢气的析出反应,可合理改善阳离子膜中带负电荷官能团的含量,实现对阳离子传导速率的调控。碱性阴离子交换膜的工作原理是负离子(例如OH-)从阴极到阳极的传导,这种离子传导机制比阳离子交换膜更适合CO2RR,不仅避免了HER反应,还促进CO2到产物的正向反应,但CO2也会与传导来的OH-发生反应,生成HCO3-或CO32-,HCO3-或CO32-会穿透阴离子交换膜到阳极造成碳损失,因此针对此问题,可调控膜的厚度、引入季铵盐官能团或填充剂(如TiO2、SiO2、ZrO2)等。 总之,电催化CO2还原是一个非常有前景的研究课题,但要实现工业化和产业化还需要解决很多问题,特别是催化剂的长时间稳定性。因此需要继续进行高性能催化剂的开发,从经济的角度思考,还需要对反应装置的结构进行优化,同时针对不同的产物还要开发新型的电解液,且要考虑反应条件的限制,另外中间隔膜也会影响反应的性能。4.3 阴离子的影响
5 反应器和隔膜研究进展
5.1 传统H槽式反应器
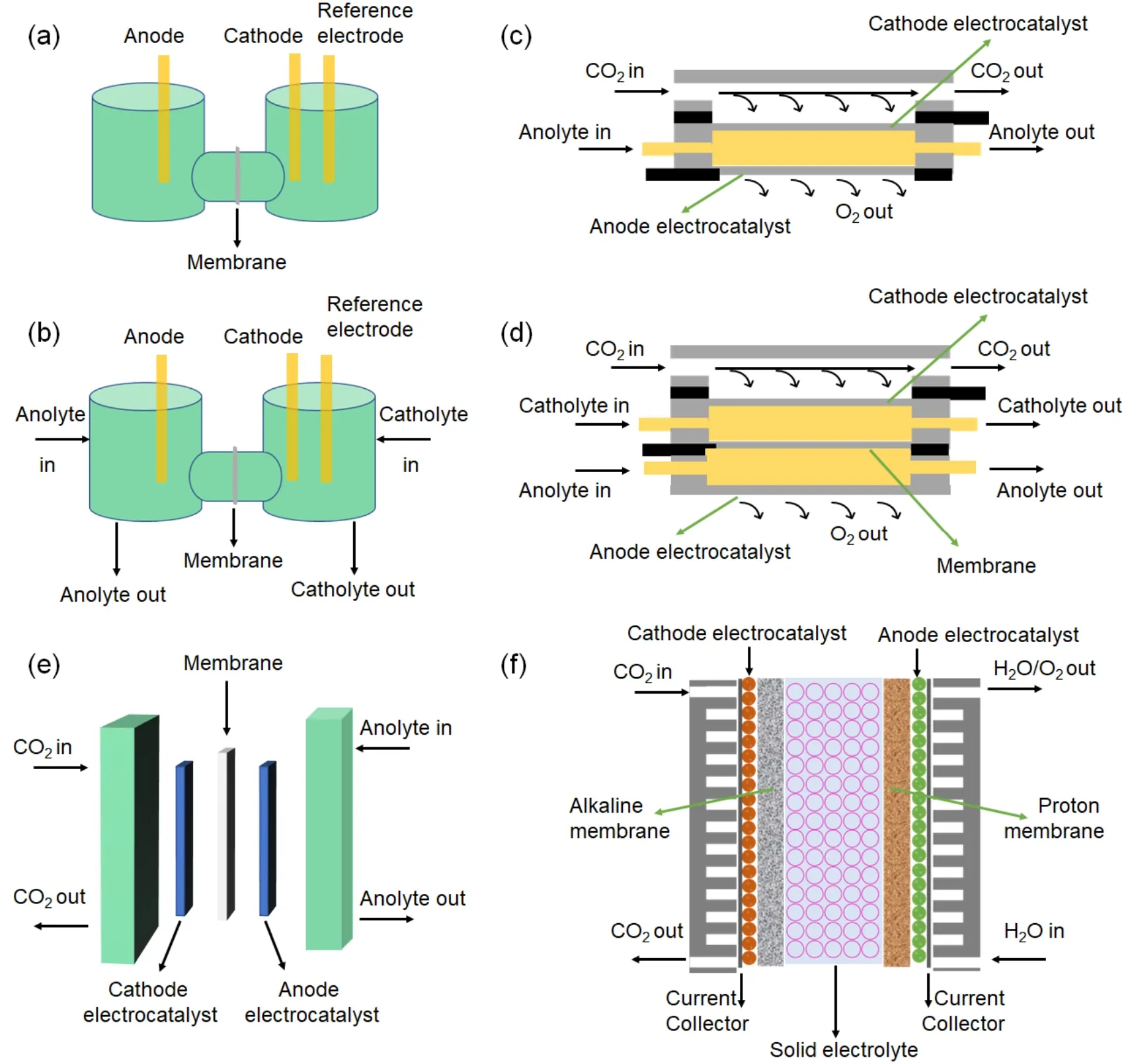
5.2 基于H型的流动式反应器
5.3 微流控反应器
5.4 气腔室反应器
5.5 膜基反应器
5.6 三腔室反应器
5.7 膜的类型

6 总结与展望
猜你喜欢
杂志排行

物理化学学报的其它文章
- Introducing Novel,Multiple Cd Coordination Modes into Gold Nanoclusters by Combined Doping for Enhancing Electrocatalytic Performance
- 用于高灵敏快速核酸检测的荧光碳点
- 利用多氟丙烯酸酯添加剂提升准二维钙钛矿发光二极管性能
- Recent Advances in Self-Supported Transition-Metal-Based Electrocatalysts for Seawater Oxidation
- 高效光电调控钙钛矿量子点阻变存储性能
- 电子自旋效应在电催化剂中的作用