黑曲霉尿苷/尿嘧啶营养缺陷型转化系统的构建及应用
2024-01-18王小平王升帆梁玲玲许新德郑建永
王小平,宋 问,张 霏,刘 燕,王升帆,邵 东,梁玲玲,许新德,郑建永
(1.浙江可明生物医药有限公司,浙江 新昌 312500;2.浙江工业大学 生物工程学院,浙江 杭州 310014;3.浙江医药股份有限公司新昌制药厂,浙江 新昌 312500)
丝状真菌黑曲霉(Aspergillusniger)广泛存在于自然界中,它在食品与发酵工业领域有着举足轻重的位置。黑曲霉具备强大的蛋白分泌表达和结构修饰能力,被称为制备柠檬酸、抗生素和酶制剂的生物细胞蛋白工厂,目前被广泛应用于食品、药品、农业、工业和医疗等行业中[1]。黑曲霉作为一种食品级的安全生产菌株,已得到美国食品和药品管理局的正式批准[2]。随着现代生物的发展,黑曲霉领域取得了许多技术进步,转化效率和目标蛋白的表达量均有一定程度的提高。其中,一个新的基因组标记手段和基因组编辑工具箱CRISPR-Cas9(成簇的规律间隔的短回文重复序列关联蛋白技术)系统被进一步研究开发[3],实现了黑曲霉基因组中的基因敲除、基因插入、碱基编辑、启动子替换和基因表达调控等[4-5]。然而,相较于大肠杆菌和枯草芽孢杆菌,黑曲霉生长周期更长,转化子的验证和纯化子的稳定遗传也较为复杂。如果要建立一个良好的黑曲霉遗传转化体系,必然离不开一个快速且高效的筛选标记[6-7]。目前,黑曲霉中的常见筛选标记主要有3大类:1) 药物抗性筛选标记,主要是抗性基因(潮霉素抗性[8]、博来霉素抗性[9]和G-418硫酸盐抗性等[10]),这是一种根据转化子耐药性的筛选方法;2) 功能性筛选标记,较为常见的是营养缺陷互补型筛选标记(尿嘧啶营养缺陷型[11]、色氨酸营养缺陷型[12]等),即构建相应的营养缺陷型宿主,再向其转入互补基因来恢复野生型表现,从而达到筛选阳性转化子的目的;3) 可视化筛选标记,通过加入报告基因(红色荧光蛋白[13]和增强型绿色荧光蛋白[14]等)达到一定的可视化效果,进而实现定性和定量的分析。
笔者利用CRISPR/Cas9技术构建了一株敲除黑曲霉CCTCC 206047乳清酸核苷-5′-单磷酸脱羧酶基因(pyrG)并能稳定遗传的黑曲霉尿苷/尿嘧啶营养缺陷型菌株。尿苷/尿嘧啶营养缺陷型中pyrG基因编码乳清酸核苷-5′-磷酸脱羧酶,对尿嘧啶核苷的合成起重要作用。诱变剂5-氟乳清酸(5-FOA)通过进入嘧啶合成途径来抑制细胞增殖,它先被转化为嘧啶类似物,进而被转化为尿嘧啶核苷合成中的自杀抑制剂5-氟尿嘧啶(5-FUMP)[15]。当培养基中含有5-FOA和尿嘧啶核苷时,正常菌株优先利用5-FOA,进而转变成有毒的5-FUMP,导致菌株死亡。若该基因发生突变或被敲除,菌株直接利用培养基中添加的尿嘧啶核苷,因而可以正常生长。利用这一特性,还可以实现培养基的反向筛选,通过不添加尿嘧啶核苷的培养基筛选pyrG基因回补表达菌株。利用流式细胞仪的分选功能,筛选得到表达绿色荧光蛋白基因的工程菌。pyrG遗传转化体系的构建极大地简化了黑曲霉转化子的筛选流程,彰显了报告基因和营养缺陷型标记在菌株基因改造中的巨大潜力。
1 材料与方法
1.1 材 料
1.1.1 菌株与质粒
黑曲霉CCTCC 206047、原核表达载体pET-28a(+)由本实验室保藏;大肠杆菌感受态细胞E.coliDH5α购自杭州擎科新业生物技术有限公司;质粒pCAS_gpyrG1购自addgene网站(www.addgene.org/)。实验构建质粒:pyrG敲除质粒P1-pyrGLR;pyrG回补质粒P2-pyrGYS;绿色荧光蛋白表达质粒P3-pyrGYS-gpdA-egfp。
1.1.2 实验试剂
PrimeSTAR®Max DNA Polymerase、QuickCutTMDpnⅠ,购自TakaRa公司;2×T5 Super PCR Mix,购自Tsingke公司;EasyPure Plasmid MiniPrep Kit,EasyPure PCR Purification Kit,购自TransGen公司;Ezup柱式真菌基因组DNA抽提试剂盒,购自Sangon公司;其他常规试剂及药品为国产或进口分装。
1.1.3 培养基和溶液
DPY培养基:酵母粉5 g/L,蛋白胨10 g/L,葡萄糖20 g/L,KH2PO45 g/L,MgSO4·7H2O 0.5 g/L;PDA培养基:称取200 g已削皮的马铃薯,充分煮沸,滤网过滤所得滤液中加入20 g葡萄糖,冷却后定容至1 L,加入质量分数为2%的琼脂粉,pH自然,115 ℃灭菌30 min;高渗CD固体培养基:蔗糖350 g/L,KCl 2 g/L,KH2PO41 g/L,MgSO4·7H2O 0.5 g/L,FeSO4·7H2O 0.01 g/L,NaNO33 g/L,琼脂20 g/L;高渗CD软琼脂培养基:成分同上,琼脂质量浓度为5 g/L。以上培养基需高温高压灭菌,不含葡萄糖的培养基在高压蒸汽锅中121 ℃条件下灭菌20 min。
STC溶液:山梨醇10.93 g,Tris 0.303 g,无水CaCl20.28 g定容于50 mL超纯水,pH自然,过膜除菌,4 ℃储存;PEG缓冲液:PEG 6000粉末15 g,Tris 0.03 g,无水CaCl20.14 g定容于25 mL超纯水,pH自然,过膜除菌,4 ℃条件下储存;尿嘧啶核苷:10 g尿嘧啶核苷溶于10 mL超纯水,过膜除菌,于-20 ℃条件下保存;潮霉素B(hygB):称取0.5 g hygB溶于10 mL无菌ddH2O,0.22 μm滤膜过滤除菌并分装,于-20 ℃条件下避光保存;0.8 mol/L NaCl溶液:NaCl 46.8 g,加入ddH2O溶解并定容至1 L;酶解液:0.2 g纤维素酶,0.1 g蜗牛酶,0.1 g溶壁酶,0.05 g溶菌酶,0.468 g NaCl,1 mL上述磷酸钠缓冲液,加入9 mL ddH2O充分溶解,0.22 μm滤膜过滤除菌,于4 ℃条件下保存。
1.2 实验方法
1.2.1 转化质粒和片段的构建
根据表1中的引物,以黑曲霉CCTCC 206047基因组为模板,用引物Up-pyrG-F/Up-pyrG-R,Do-pyrG-F/Do-pyrG-R分别扩增pyrG基因(Gene ID:4985706)带有同源臂的上游(1 407 bp)和下游(1 410 bp)基因片段。纯化后的pyrG上下游片段互为引物和模板进行融合PCR,然后用引物pyrG-F/pyrG-R扩增pyrGLR片段(2 817 bp)。以质粒pET-28a(+)为模板,用引物28ZT-F,28ZT-R反向扩增载体片段(5 356 bp),通过一步克隆试剂盒组装成质粒P1-pyrGLR;使用引物pET-28a(+)-egfppyrG-F/pyrG-R扩增pyrGYS(包含pyrG基因、上游启动子和下游终止子片段),与载体pET-28a(+)连接后构建质粒P2-pyrGYS;以含gpdA启动子(2 132 bp)和egfp基因(714 bp)的质粒为模板,使用引物28ZT-egfp-F/28ZT-R扩增载体片段,然后通过一步克隆试剂盒组装成回补表达质粒P3-pyrG-gpdA-egfp。以上述3个质粒为模板,分别扩增pyrGLR(2 781 bp),pyrGYS(3 908 bp)和pyrGYS-egfp(4 236 bp)片段,用于后续黑曲霉原生质体的转化。

表1 本实验用到的引物
1.2.2 黑曲霉基因组的提取
挑取培养基上新鲜的黑曲霉孢子,加入50 μL缓冲液Buffer digestion后用电动手持研磨器研磨2 min,补足200 μL缓冲液Buffer digestion后加入20 μL Protein K和2 μLβ-巯基乙醇,混匀后56 ℃水浴1 h,使菌体组织充分裂解,然后使用Ezup柱式真菌基因组DNA抽提试剂盒提取转化子基因组,具体操作详见试剂盒使用说明。
1.2.3 黑曲霉转化子的筛选
通过PEG介导原生质体转化法将pyrG基因的上下游同源臂融合片段转化到黑曲霉CCTCC 206047原生质体中。转化实验组的高渗CD培养基中需要添加尿嘧啶核苷和5-FOA作为筛选条件。30 ℃正置培养3~5 d,挑取平板上长出来的转化子,转移至含有5-FOA和尿嘧啶核苷的PDA平板中,获得第一代转化子。经基因组验证正确的转化子在对应平板传代数次后,获得一株稳定遗传的营养缺陷型宿主。将pyrGYS和pyrGYS-egfp基因片段通过PEG介导原生质体转化法导入到营养缺陷型宿主菌中,阳性转化子可恢复野生型表型,使用基础PDA培养基即可初步筛选。
1.2.4 5-FOA最小抑制浓度的确定
根据pyrG基因反向筛选的特性,通过在培养基中添加5-FOA来筛选野生型黑曲霉。具体的5-FOA用量(即对黑曲霉的最小抑制浓度)仍需考察。用已灭菌的移液器10 μL吸头将野生型黑曲霉接种在含不同质量浓度5-FOA(0,0.5,1.0,1.5,2.0,3.0 g/L)的培养基中,30 ℃培养3 d,观察并记录其生长抑制情况。
1.2.5 黑曲霉转化子的遗传稳定性
将在筛选平板中生长和基因组测序正确的转化子接种到含5-FOA和尿嘧啶核苷的PDA平板中,在30 ℃恒温培养箱中正置培养3~5 d,观察其生长状况,挑取生长良好的转化子继续传代培养,以观察其性状的遗传稳定性。
1.2.6 黑曲霉转化子的抗性丢失
结合杨艳坤等[16]报道的菌株质粒丢失的高通量筛选方法,将含有抗性的黑曲霉营养缺陷型菌株进行抗性基因丢失处理。首先,对黑曲霉阳性转化子进行稀释涂布,挑取单菌落溶于无菌ddH2O中,经超声处理后制成相对均一的孢子悬液;然后,接种于含5-FOA和尿嘧啶核苷的DPY培养基中,30 ℃,200 r/min条件下摇床培养4 h;最后,分别稀释涂布于含hygB和不含hygB的筛选培养基中,30 ℃正置培养3~4 d。在含hygB的筛选培养基中无法生长,而在不含hygB筛选培养基中可以生长的菌株即为黑曲霉营养缺陷型Cas9质粒丢失菌株。
1.2.7 流式细胞仪FCM分选黑曲霉单孢子
将绿色荧光回补表达质粒转入营养缺陷型宿主后,菌株将恢复合成尿苷能力,即菌株不仅能在基础PDA培养基中正常生长,而且能表达绿色荧光蛋白,因而可用流式细胞仪分选转化子。用PBS缓冲盐溶液洗下在PDA转化板中生长的黑曲霉孢子,用神奇滤布过滤后收集孢子,洗涤3次,稀释至OD为0.3~0.5。以黑曲霉营养缺陷菌株的孢子悬液样品作为阴性对照,样品由流式细胞仪(BD FACSMelody)FITC-Log通道检测。预先在48孔板中加入700 μL固体PDA培养基,将荧光信号强的单个孢子分选入单独的孔中,30 ℃正置培养3~5 d。
1.2.8 转化子的荧光测定
用EP管加入适量无菌水,挑取PDA培养基中适量的新鲜孢子进行稀释。以营养缺陷型黑曲霉宿主菌的孢子悬液作为对照。准备干净的载玻片,用移液器吸取10 μL孢子悬液滴在载玻片上,盖上盖玻片,轻轻按压,挤去气泡和多余的无菌水。将载玻片放置在荧光显微镜(EVOS M5000)的载物台上,在20倍镜明场下调节至清晰状态,切换光源观察样品是否激发出绿色荧光。使用荧光成像系统(470/525 nm)进行检测,以20倍物镜拍摄荧光图像。
2 结果与分析
2.1 利用CRISPR/Cas9技术敲除pyrG
近年来,通过同源重组将外源DNA靶向整合至黑曲霉和其他丝状真菌基因组中的方法得到广泛应用[17]。研究表明DNA双链断裂(DSB)能提高同源重组效率。现在普遍存在的CRISPR/Cas9便是为快速靶向DSB而设计的[18]。在本研究中,利用质粒pCAS_gpyrG1表达Cas9蛋白和pyrG-SgRNA序列,通过Cas9蛋白引导pyrG-SgRNA序列到黑曲霉基因组pyrG位点[19],实现基因组编辑,从而达到敲除pyrG的目的。利用CRISPR/Cas9技术敲除pyrG的原理如图1所示。

图1 利用CRISPR/Cas9技术敲除黑曲霉pyrG示意图Fig.1 Schematic diagram of principle of knock out of Aspergillus niger pyrG using CRISPR/Cas9 technology
2.2 pyrG基因敲除菌株的筛选及验证
参照Boeke等[20]为酿酒酵母开发的突变体的阳性选择法来筛选黑曲霉的pyrG突变体。该方法基于pyrG缺陷型突变体在尿嘧啶或尿苷存在下对5-FOA的抗性。由于pyrG基因编码乳清酸核苷-5′-单磷酸脱羧酶,能够将无毒的5-FOA催化为毒性产物5-FUMP,故尿苷/尿嘧啶营养缺陷型转化子能在含有5-FOA的培养基中正常生长,而未破坏/敲除pyrG的宿主菌无法生长。黑曲霉转化子和宿主菌株在筛选培养基中的生长情况如图2所示。由图2可知:黑曲霉阳性转化子(a,b,c区域)可以在含有5-FOA和尿苷的复筛培养基中正常生长,而宿主菌株(d区域)无法生长。
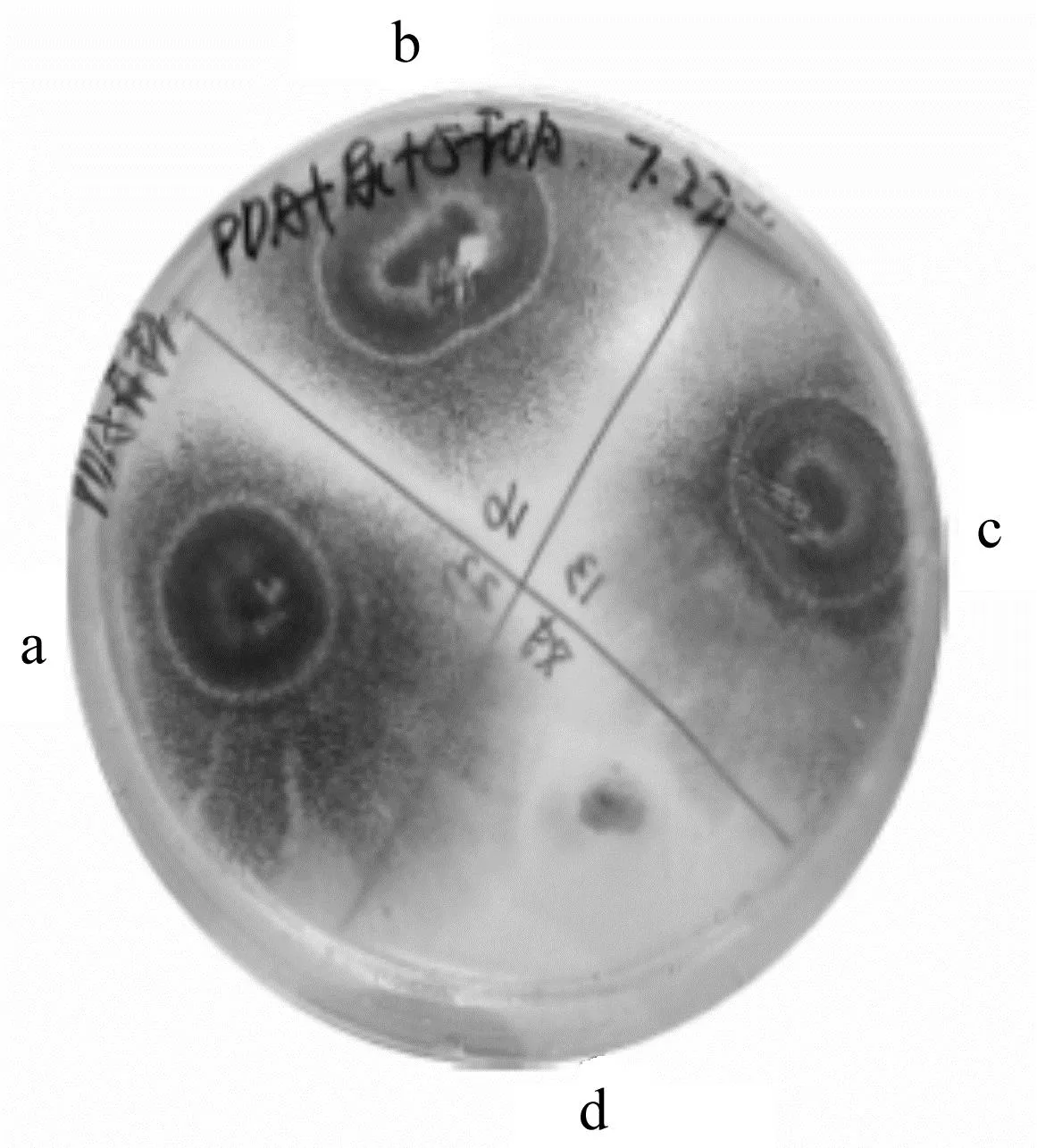
图2 黑曲霉转化子和宿主菌株在筛选培养基中的生长情况Fig.2 Growth of Aspergillus niger transformants and host strains in screening medium
提取在筛选培养基上表型正常的黑曲霉转化子的基因组,用引物YZ-1,YZ-2进行基因验证,采用核酸电泳检测。引物YZ-1,YZ-2位于pyrG左右同源臂上,阳性转化子和阴性转化子PCR扩增条带应相差1 127 bp。黑曲霉中荧光缺陷型转化子基因组PCR电泳结果如图3所示。图3中:泳道control为宿主对照菌株对照组;泳道1,2,3,4为敲除pyrG菌株样品组;泳道M为核酸分子量标准品2K plus Ⅱ Marker;泳道5,6的PCR产物条带单一且表观分子质量与理论分子质量相符,将其送去测序。实验结果表明利用CRISPR/Cas9技术可以实现黑曲霉CCTCC206047中pyrG基因的成功敲除。

图3 黑曲霉中荧光缺陷型转化子基因组PCR电泳结果Fig.3 PCR electrophoresis results of transformant genome in Aspergillus niger
2.3 黑曲霉转化子的抗性基因丢失
对测序正确的黑曲霉营养缺陷型菌株进行抗性丢失处理,结果如图4所示。在含潮霉素B的筛选培养基中无法生长,而在不含潮霉素B筛选培养基中可以生长的菌株即为黑曲霉营养缺陷型Cas9质粒丢失菌株。将黑曲霉营养缺陷型转化子命名为PA-1,随后进行pyrG基因回补表达。
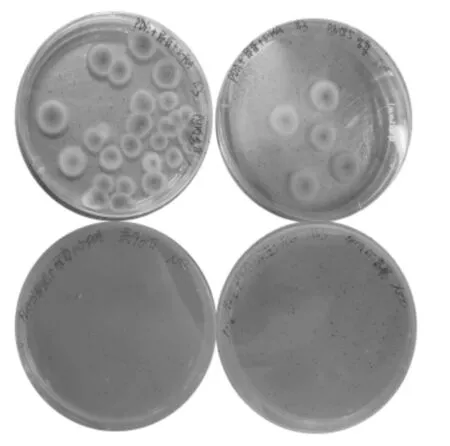
图4 转化子在含或者不含hygB筛选培养基中的生长情况Fig.4 Growth of transformants in screening media containing or without hygB
2.4 5-FOA最小抑制质量浓度的确定
5-FOA自身对黑曲霉细胞没有毒性,然而黑曲霉宿主表达的pyrG酶能够将5-FOA催化为有毒性的5-FUMP,从而杀害自身细胞。观察黑曲霉在含有不同质量浓度的5-FOA培养基中的生长情况,从而确定达到抑制效果的5-FOA最小质量浓度。5-FOA最小抑制质量浓度如表2所示。表2中:“+”代表黑曲霉宿主可以生长;“-”代表黑曲霉宿主不可以生长)。由表2可知0.75 g/L的5-FOA不能完全抑制黑曲霉宿主的生长。考虑到5-FOA的成本和抑制效果,后续实验选择5-FOA质量浓度为1.0 g/L。
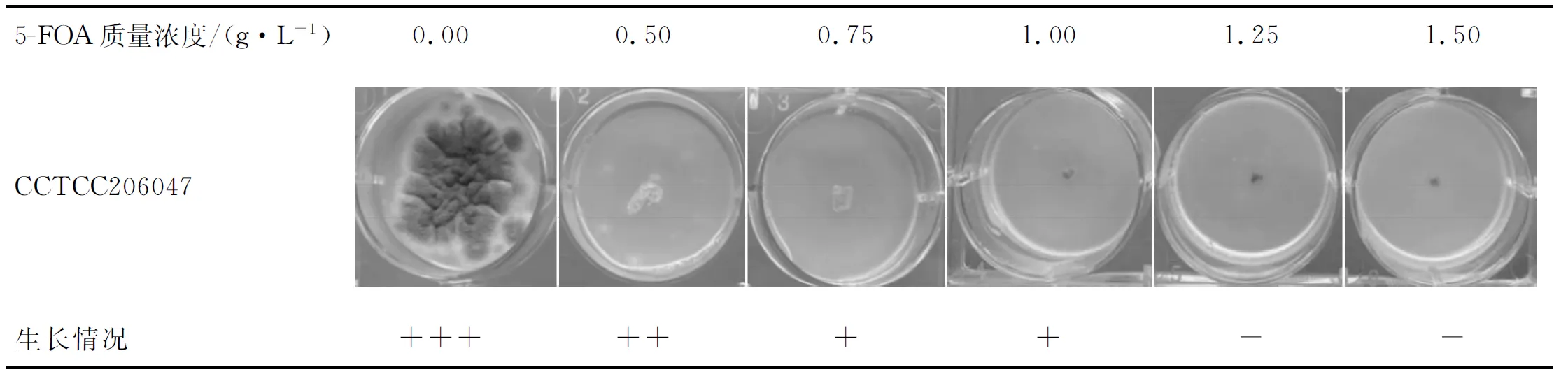
表2 5-FOA最小抑制质量浓度
2.5 黑曲霉转化子的遗传稳定性
挑取测序正确的黑曲霉阳性转化子PA-1在筛选培养基中进行传代培养,考察其遗传稳定性。筛选培养基为含有5-FOA、尿嘧啶核苷的PDA培养基。黑曲霉转化子的遗传稳定性如图5所示。由图5可知:阳性转化子经10代遗传后在筛选培养基中长势良好,进而验证了敲除pyrG基因的这一性状在黑曲霉转化子中能够保持稳定的遗传。
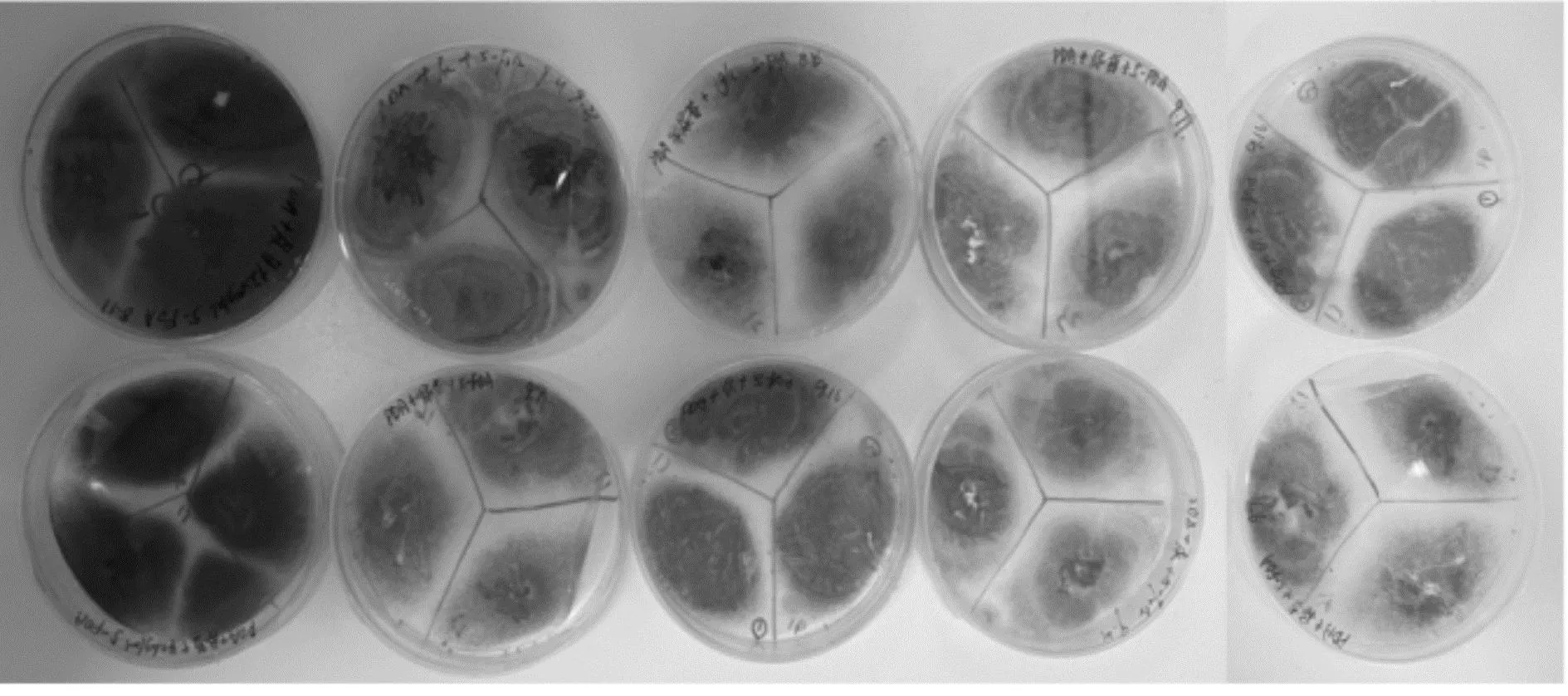
图5 黑曲霉转化子的遗传稳定性Fig.5 Genetic stability of Aspergillus niger transformants
2.6 黑曲霉营养缺陷型菌株的回补表达
为了考察pyrG基因的有效性,将pyrG基因重新导入尿苷/尿嘧啶营养缺陷型菌株PA-1中,测试转化后菌株的表型。回补表达黑曲霉转化子的验证结果如图6所示。由图6(a)可知:pyrG回补后的黑曲霉菌株可以在不添加尿嘧啶核苷和5-FOA的基础PDA培养基中生长。使用引物YZ-3,YZ-4进行PCR,验证提取转化子的基因组中是否含有pyrG基因。由图6(b)可知:泳道2,5,6,7,10的黑曲霉转化子的表观分子质量与理论分子质量2 310 bp相符。实验证明了从黑曲霉CCTCC206047基因组中扩增得到的pyrGYS回补标记可用于pyrG型尿嘧啶/尿嘧啶核苷营养缺陷型黑曲霉恢复突变,同时证明黑曲霉PA-1是一株可进行基因转化的pyrG缺陷型菌株。
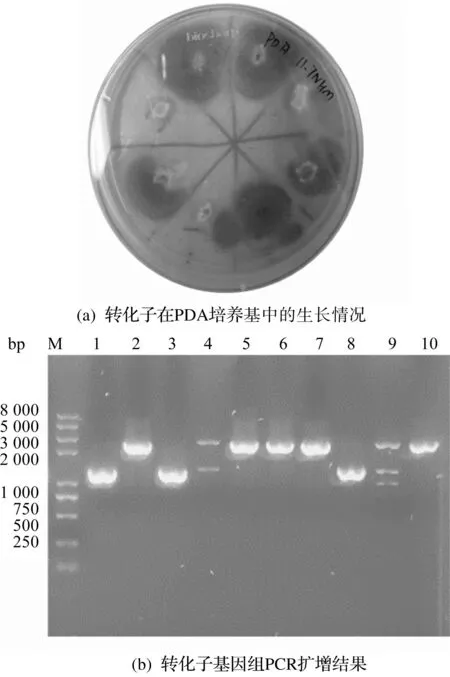
图6 回补表达黑曲霉转化子的验证Fig.6 Validation of complementary expression in Aspergillus niger transformants
2.7 黑曲霉转化子荧光表达情况
增强型绿色荧光蛋白EGFP是GFP优化后的衍生物,具有更强的荧光信号,已成为评估遗传转化系统的重要工具。黑曲霉分生孢子的荧光表达情况如图7所示。由图7可知:与没有荧光的亲本菌株PA-1相比,egfp基因回补表达转化子PG-gpdA的分生孢子有较为明显的荧光现象。实验结果表明:黑曲霉回补pyrG基因的同时能实现目标基因egfp的高效表达,可进一步通过流式细胞仪进行定量分析。

图7 黑曲霉分生孢子的荧光表达情况Fig.7 Fluorescence expression of Aspergillus niger conidia
2.8 流式细胞仪的分选
利用流式细胞仪分析egfp基因回补表达黑曲霉菌株PG-gpdA,通过流式细胞仪的分选功能,将荧光信号强的单孢子分选到48孔板中。流式细胞仪的分选情况如图8所示。由图8可知:PDA平板中的10 000个孢子中约有12.4%的孢子能表达egfp;流式细胞仪分选后的单孢子在48孔板上存活率达85.4%;随机挑选基础培养基上生长的转化子到荧光显微镜下进行观察,均能观察到较为明显的荧光信号。
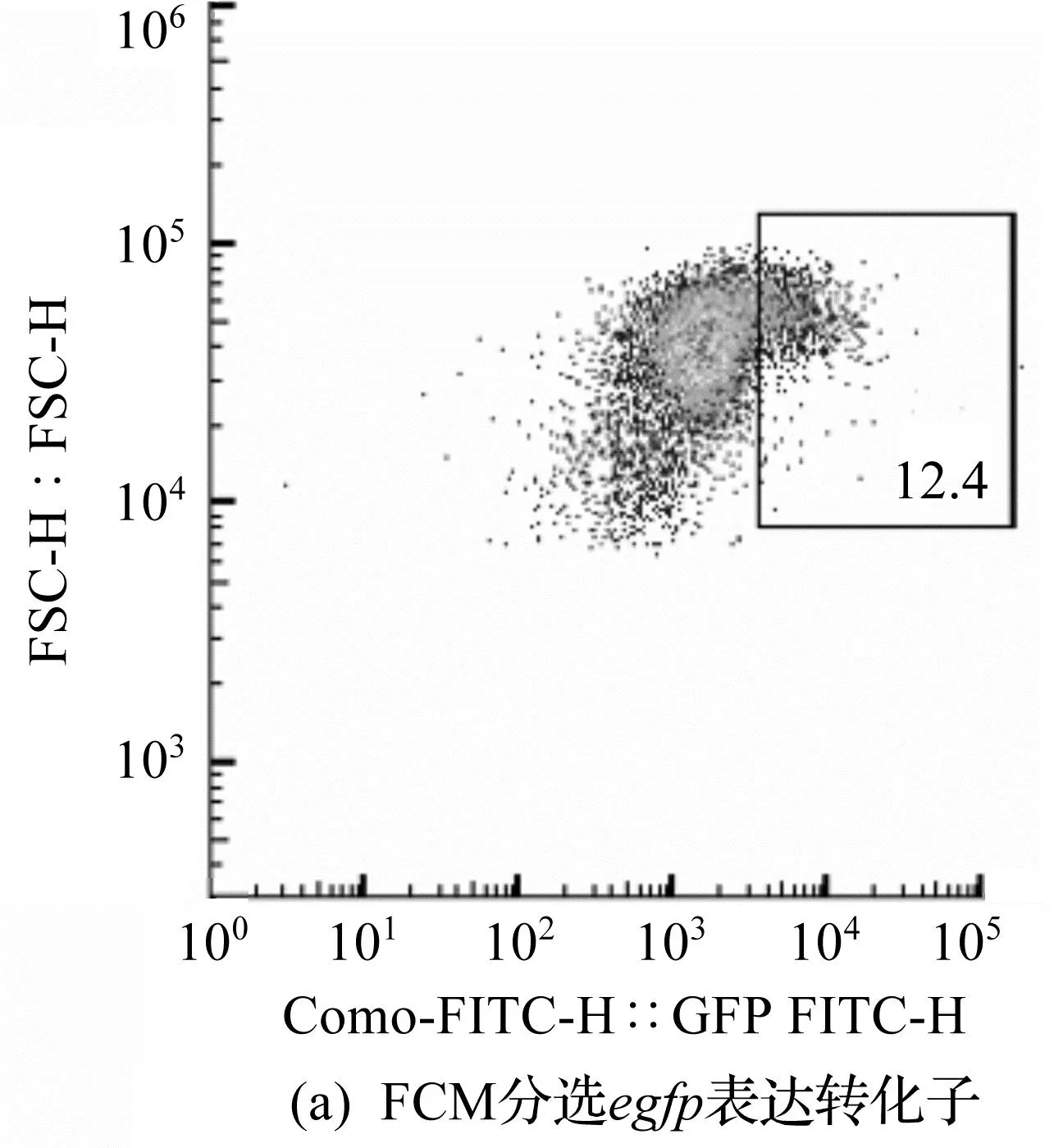
图8 流式细胞仪的分选情况Fig.8 Flow cytometry sorting diagram
3 结 论
以黑曲霉CCTCC 206047基因组为模板,利用CRISPR/Cas9系统实现pyrG基因的重组敲除,在含有5-FOA、尿嘧啶核苷和潮霉素B的培养基上筛选表型正确的转化子。通过考察黑曲霉转化子的遗传稳定性和基因组PCR扩增验证,进一步获得一株成功敲除黑曲霉pyrG基因的阳性转化子,丢失抗性质粒后即为黑曲霉pyrG营养缺陷型菌株PA-1,经10代遗传后转化子在筛选培养基中长势良好,验证了敲除pyrG基因的这一性状能够稳定遗传。将包含黑曲霉pyrG基因表达框的重组质粒P2-pyrGYS转化至营养缺陷型菌株PA-1中,恢复其野生型表型,进而验证该营养缺陷型系统的可行性。利用该遗传转化系统可实现egfp基因的重组表达,通过营养筛选和流式细胞术为黑曲霉转化子提供双重检测指标。该筛选办法简化了黑曲霉阳性转化子的筛选流程,建立了一个快速的可视化的高通量筛选模型。然而该检测办法仍处于孢子水平,后续目的基因的表达仍受分泌途径和菌丝形态的调控,仍需建立更加精确的黑曲霉筛选技术。
