OXCT1基因缺陷致琥珀酰辅酶A:3-酮酸辅酶A转移酶缺乏症1例并文献复习
2024-01-07陈俊龙王小丽李俊利张晓梅
陈俊龙, 王小丽, 李俊利, 张晓梅
(内蒙古自治区人民医院, 内蒙古 呼和浩特 010017)
琥珀酰辅酶A:3-酮酸辅酶A转移酶(Succinyl-CoA:3-ketoacid CoA transferase,SCOT),基因位点标记OXCT1,EC 2.8.3.5,存在于多个组织的线粒体内,能将乙酰乙酸转化为乙酰乙酰辅酶A,乙酰乙酰辅酶A在乙酰乙酰辅酶A硫解酶(T2)的作用下分解为乙酰辅酶A后进入三羧酸循环。该酶是酮体代谢过程中的关键限速酶。先天性SCOT缺乏为常染色体隐性遗传病,1972年J.T.Tildon等首次进行了报道[1]。现对1例确诊的OXCT1基因突变导致的先天性SCOT缺乏病例进行分析总结,并通过复习相关文献归纳总结其特征,以便提高临床工作中对该病的认识,达到及时确诊、早期治疗的目的。
1 病例资料
患儿女,5月龄,因“呼吸困难1小时”就诊,起病急,入院前1h无明显诱因出现喘憋,面色发灰,伴有呻吟、烦躁不安,并呈进行性加重。患儿系第2胎第2产,胎龄39周选择性剖宫产分娩,母孕期健康,无窒息史,出生体重3.1kg,出生身长51cm,分别为同年龄、同性别儿童的第48百分位(48th percentile,P48)、P58。纯母乳喂养,3月龄体检时体重6.1kg,身长60cm,分别为同年龄、同性别儿童的P52、P51,0~6岁儿童神经心理发育量表评估智龄为3月龄,父亲患有慢性乙肝病毒感染,未规范治疗,母亲身体健康,非近亲结婚,有一姐姐,6岁,健康,否认家族性遗传病及传染病史。
体格检查:体重7.4kg,身长66cm,头围42cm,分别为同年龄、同性别儿童的P50、P60、P54,反应差,急性病面容,前囟未闭,大小3.0cm×3.0cm,颈抵抗(-),呼吸困难明显,三凹征(+),双肺呼吸音粗,未闻及湿罗音,心腹未见异常,四肢肌力、肌张力均正常,双侧膝腱反射、巴宾斯基征对称引出。
辅助检查及诊疗经过:入院时血气分析PH7.04、BE-24.5mmoL/L、AG 33mmoL/L、Lac1.8mmoL/L,给予CPAP无创通气、碳酸氢钠纠酸等对症治疗后呼吸困难明显缓解,撤无创呼吸机继续抗感染、营养心肌巩固治疗,入院第4天再次出现严重呼吸困难,复查血气分析提示:PH7.13、BE -24.2mmoL/L、AG 27mmoL/L、Lac1.1mmoL/L,再次予以纠酸治疗后呼吸困难缓解,继续巩固对症治疗;期间完善辅助检查提示血清肌酸激酶、肌酸激酶同工酶及乳酸脱氢酶均轻度升高,血糖5.02mmoL/L,肝肾功、胰岛素、电解质均正常,心彩超及全腹彩超均未见明显异常,血氨109mmoL/L(参考范围9~33mmoL/L),纠酸治疗中多次复查血气分析均提示高AG代谢性酸中毒,多次尿常规均提示酮体2+~4+,经积极治疗出院前复查尿酮体3+,考虑患儿不明原因严重酮症酸中毒,建议其完善血尿代谢及染色体检查明确,家属拒绝,临床症状好转出院;
患儿出院回家后5d再次出现呼吸困难发作,急诊入院,入院血气分析提示PH7.18、BE-22.1mmoL/L、AG30mmoL/L、Lac1.1mmoL/L,血氨46mmoL/L,胸片提示肺炎,再次予以CPAP无创通气、反复纠酸等对症治疗后呼吸困难很快缓解,期间完善辅助检查生化提示肝肾功、电解质、心肌酶正常,血糖、糖化血红蛋白、胰岛素、C肽水平正常,血氨、PCT、CRP、TORCH、免疫球蛋白、肺炎链球菌抗原、呼吸道病毒抗体谱、血培养均未见异常。心电图窦性心律,PR间期延长。头颅CT脑外腔隙增宽,符合左额颞顶部皮下积液表现。完善血串联质谱代谢检测回报:C3DC/C4CH(丙二酰基肉碱/3-羟基丁酰基肉碱)增高;尿液有机酸检测:2-羟基丁酸2增高、3-羟基丙酸-2增高、3-羟基丁酸-2、2-甲基-3-羟基丁酸-2、3-羟基异戊酸-2、乙基羟基丙酸-2、3-羟基戊酸-2,均提示酮症,家长同意完善全外显子基因检测,但结果未回报。经积极治疗后呼吸平稳恢复正常,完善头颅MRI检查提示双侧颞叶皮层下脱髓鞘改变,余未见异常。病情稳定好转出院,回家后氨基酸配方奶粉喂养,口服左卡尼汀、赖氨基醇口服液、维生素B族、益生菌等药物,期间由于家庭条件随访依从性差,每月自行县级医院门诊复查尿常规监测尿酮体变化,未再出现酸中毒复发,直至3月后(患儿8月龄)我院门诊复查,完善儿心量表评估提示智龄为7月龄,动态脑电图监测正常,脑干听觉诱发电位双耳通过,精神运动发育正常。
2 基因检测
征得家长同意并签署知情同意书,患儿及父母亲完善了家族全外显子基因检测。发现患儿OXCT1基因(NM_000436)5号外显子41870386位点存在c.75C>A(p.Tyr25Ter)杂合无义变异,该变异来源于父亲;5号外显子41762248位点存在c.1303A>C(p.Thr435Pro)杂合错义变异,该变异来源于母亲,见图1。
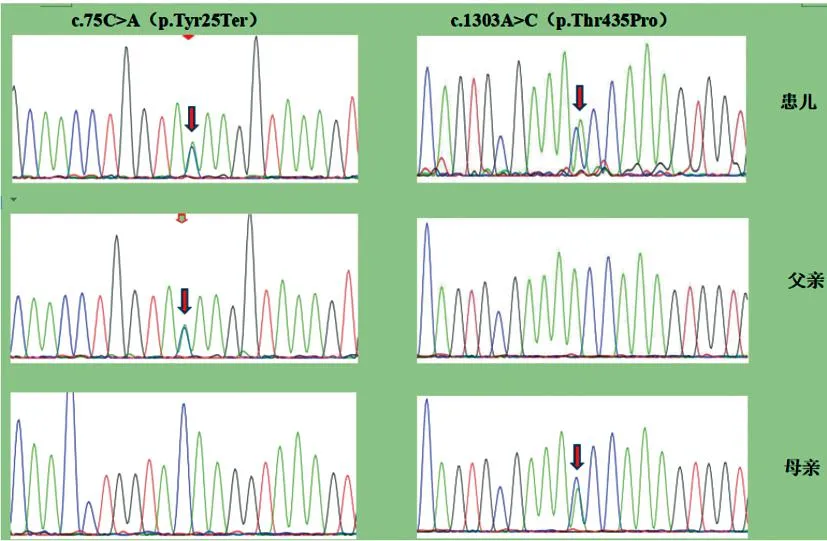
图1 患儿及其父母OXCT1基因Sanger测序结果
两种变异在外显子组整合数据库(The Exome Aggregation Consortium,EXAC)、外显子组测序计划(Exome Sequencing Project,ESP6500)、千人基因组(1000Genomes)均未收录。检索人类基因突变数据库(The Human Gene Mutation Database,HGMD)、PubMed、ClinVAr数据库发现,c.75C>A和c.1303A>C变异的致病性均未见文献报道,该变异可能为国际上未报道的新变异。
根据2015年美国医学遗传学与基因组学学会(AmericAn College of MedicAl Genetics And Genomics,ACMG)标准和指南[2],本例患儿OXCT1基因变异c.75C>A(p.Tyr25Ter)为致病性变异(PVS1+PM2+PP3),c.1303A>C(p.Thr435Pro)为疑似致病性变异(PM1+PM2+PP2+PP3)。
3 文献检索
以“琥珀酰辅酶A:3-酮酸辅酶A转移酶缺乏”、“SCOT”、“OXCT1”、“Succinyl-CoA:3-ketoacid CoA Transferase Deficiency ”为检索词,检索中国知网、万方数据库及PubMed数据库建库至2022年10月31日的相关文献并进行复习。检索到符合条件的英文文献34篇,相关中文文献2篇。剔除资料不完整病例,经整理发现通过遗传学分析确诊的SCOT缺乏症患儿共52例,国内报道4例,国外报道48例,现对已报道的52例OXCT1基因变异以及本组1例,共53例SCOT缺乏症进行总结分析。
国内4例病例中,男3例,女1例,发病时最小年龄1月,最大年龄8月,其中1例为c.1057T>G(p.Y353D)/c.1059DupT/c.1059delG复合杂合变异,另外1例为12外显子的跳跃(c.1147G>A),剩余2例均符合SCOT缺乏的临床诊断标准,即:反复发作的酮症酸中毒,无低血糖发作情况下尿酮体强阳性,尿GC-MS分析示尿中出现乙酰乙酰和3-羟基丁酸,其他实验室检查无异常,但未检测到致病突变,作者解释了这种阴性结果的出现可能与其实验用的RT-PCR克隆测序法、其他基因或非解酮障碍疾病有关[3-5]。
国外48例病例中,男25例,女23例,发病时最小年龄生后36h,最大年龄21岁。10例(10/48,20.8%)在新生儿期发病,26例(26/48,54.2%)在婴儿期发病,剩余12例(12/48,25.0%)1岁后发病。其中3例伴有新生儿低血糖的发生,3例因发病时出现严重的酮症酸中毒伴有电解质紊乱进行了持续性肾脏替代治疗(continuous renal replacement therapy,CRRT)后好转;1例患儿为在新生儿时期筛查确诊为苯丙酮尿症(Phenylketonuria,PKU),持续接受低苯丙氨酸饮食和氨基酸补充剂的治疗,但在9月龄时因反复呼吸困难合并酸中毒完善基因检测确诊;21例患儿因呼吸困难给予机械通气治疗后好转。48例确诊病例中所有患儿均以酮症酸中毒为首发症状,无无症状患者。临床表现均为急性失代偿性代谢性酸中毒(pH<7.2,BE>-20mmoL/L)、酮症(尿酮体3~4+)[6],这些临床特点均与本文报道的病例具有一致性。首次代谢性酸中毒失代偿期间报告的血糖浓度范围从无法检测到的低到明显升高(26.5mmoL/L),血乳酸和血氨浓度通常在其参考区间内[7],本文病例的血乳酸和血糖为正常,但血氨浓度明显增高。关于SCOT缺乏症的发病率在文献中却均未见报道,但Grünert等[8]人概括了自1972年SCOT缺乏被首次描述至2020年期间,世界范围内大约有44例患有SCOT缺乏症的患儿,它们均存在不同类别的基因突变。所报道的48例病例共有OXCT1基因变异位点43个,以错义变异(29/43,67.4%)为主,插入、缺失和无义变异8例(8/43,18.6%),内含子变异导致的外显子跳跃3例(3/43,7.0%),其他变异3例(3/43,7.0%)。
4 讨 论
酮体是脂肪分解的代谢产物,主要产生于肝脏,当机体葡萄糖供应有限时成为肝外器官提供能量的主要源泉。酮体利用(酮分解)中的任何酶异常(如SCOT缺乏或乙酰乙酰辅酶A硫解酶T2缺陷)都可能导致未使用的酮体堆积而发生酮症[8]。SCOT缺乏就是一种罕见的先天性酮体利用障碍性疾病,当体内酮浓度增加时进一步导致酮症酸中毒的发生[9]。
人类先天性SCOT缺乏编码的OXCT1基因定位于5p13,由17个外显子组成[1]。SCOT缺乏症患儿通常在新生儿期或婴儿期发病居多,可出现反复酮症酸中毒发作,严重时可能危及生命,但发作间歇期无症状[6]。在所有确诊的53例SCOT缺乏症患儿中,除最早的两例患儿死亡外,其余的病例在报告时仍然健康存活,而且几乎所有存活的患儿均表现为精神运动发育正常。目前该病报告的病例数量较少,个体症状可能也有所不同,包括呕吐、嗜睡、呼吸急促、以及严重酮症酸中毒引起的昏迷等[10]。本文病例中该患儿出生史及生长发育均正常,3次发病均以呼吸困难为主,经积极纠酸、抗感染、氧疗等对症治疗症状很快改善,临床表现为阴离子间隙增高的严重代谢性酸中毒,血乳酸正常但血氨增高,尿酮体增高及尿液有机酸检出大量酮体成分,初次诊疗考虑其可能为解酮障碍相关疾病,最终通过全外显子基因测序明确诊断[2]。且发现的OXCT1基因变异在国内外均未见文献报道,丰富了SCOT缺乏的基因数据库。
在确诊的SCOT缺乏患儿中,不论出现何种基因突变,其临床表现均以反复发作的酮症酸中毒为主,个别症状因突变的靶点不一致而表象不一[11]。值得注意的是,除了酮体,在SCOT缺陷患儿的血液和尿液样本中没有发现其他诊断性代谢产物[7-8]。因此,与大多数有机酸血症相比,单纯通过有机酸分析不可能进行SCOT缺陷的实验室诊断。尽管SCOT缺乏症是一种良性疾病,临床结局及预后往往较好,但酮体代谢危机也可能危及生命,甚至致命[4]。由于诊断只能通过酶学研究或基因突变分析,而酶活性的测定是繁琐的,突变分析是昂贵的,因此可能容易漏诊。目前SCOT缺乏症尚没有特异而有效的治疗方案,临床治疗以对症支持措施为主,如给予纠酸、呼吸支持、低蛋白低脂饮食、长期随访等来改善症状,延缓病情发展,对于确诊患儿建议定期检查尿液中的酮体,以便在重度酮尿的情况下启动早期治疗。另外可通过避免禁食和服用碳水化合物来抑制酮生成,口服碳酸氢钠、蛋白质限制性饮食和肉碱补充被建议作为辅助治疗。由于SCOT导致酮体分解障碍,基于目前已知的SCOT晶体结构入手,开发抗人SCOT抗体将其用于精确评估残余活性有望成为将来治疗本病的研究方向[7]。同时根据遗传学分析结果,在受累家庭中开展遗传咨询并对高风险胎儿进行产前诊断,是避免本病患儿出生的有效手段。
综上所述,本研究确诊的SCOT缺乏症患儿OXCT1基因变异c.75C>A(p.Tyr25Ter)及c.1303A>C(p.Thr435Pro)为新发变异,通过患儿临床、实验室及遗传学特征分析与已知世界范围内报道的SCOT缺乏确诊病例对比,对提高本病患儿的早期诊断和临床管理水平具有一定的参考价值。当生长发育正常的婴幼儿反复出现难以解释的酮症酸中毒,而临床检查无显著异常时应该考虑酮体分解障碍相关性疾病,同时也应将SCOT缺乏症纳入复发性代谢性酸中毒发作患儿的鉴别诊断中。
