基于紫精配体的新型铀酰配位聚合物的合成、结构和光致变色性能
2023-12-29李星君胡孔球于吉攀石伟群
李星君,胡孔球,梅 雷,于吉攀,张 强,石伟群
1.苏州科技大学 化学与生命科学学院,江苏 苏州 215009;2.中国科学院 高能物理研究所,北京 100049

在CPs的合成过程中,有机配体的选择是关键的一步。Thuéry等[20]研究发现,相较于简单阴离子配体和一些中性螯合配体,具有多羧酸结构的两性离子配体与铀酰离子同样易发生配位行为,因此可以应用于构建具有高聚合分子框架结构的铀酰配位聚合物。除此之外,新型功能性铀酰配合物的构建还要依靠功能基团的引入或相关活性位点的修饰。紫精类化合物[21]是一种常见的具有光活性的材料,在不同外部刺激(光照、加热、压力等)下可以导致材料的理化性质发生变化,如荧光发射、光致变色现象[22-25]、UV-vis[26]的特定吸收等。在前期工作中,本课题组[27-29]设计合成了一系列基于紫精配体的4f/5f配位聚合物,并深入研究了它们的结构和性质。例如,本课题组基于一种紫精二羧酸配体合成了两例具有明显光致变色性质的铀酰配位聚合物[27]。此外,还利用稀土离子和一种紫精四羧酸配体1,1′-双(3,5-二羧基苯基)-4,4′-联吡啶合成了两例具有稳定紫精自由基的金属-有机框架(metal-organic frameworks, MOFs)材料,二者均可以实现光催化条件下氮气的还原[28]。最近,本课题组通过逐步合成策略制备了一例基于紫精配体的稳定自由基锕系MOF材料,其在可见-近红外范围内表现出良好的光催化CO2还原能力[29]。
本工作拟以紫精衍生物1-(4-羧基苯基)-4,4′-联吡啶氯化物(Hhpcl·Cl)作为功能性配体,以邻苯二甲酸(H2PA)、间苯二甲酸(H2IPA)、对苯二甲酸(H2TPA)为辅助配体,以硝酸铀酰为金属离子源,通过水热法合成三种含有紫精基团的铀酰配位聚合物,即(UO2)(PA)(hpcl)(配合物1)、(UO2)(IPA)(hpcl)(配合物2)、(UO2)(TPA)-(hpcl)·(H2O)3·(H2TPA)0.5(配合物3),并研究它们的光化学行为。
1 实验部分
1.1 试剂与仪器
硝酸铀酰(UO2(NO3)2·6H2O),纯度99%,中国科学院高能物理研究所;对苯二甲酸、邻苯二甲酸、间苯二甲酸,纯度99%,北京伊诺凯科技有限公司;4,4-联吡啶,纯度99%,上海泰坦科技有限公司;4-氨基苯甲酸,纯度99%,国药集团化学试剂有限公司;乙腈、三乙胺,纯度99%,天津市致远化学试剂有限公司。
Bruker D8-Venture X射线单晶衍射仪、Bruker D8 Advance衍射仪、Bruker Tensor 27红外光谱仪、Bruker E-500型电子顺磁共振谱仪,德国Bruker公司;TA Q500分析仪,美国TA仪器公司;F-4500型荧光分光光度计,天美(中国)科学仪器有限公司;U-3900型紫外可见分光光度计,日本日立公司。
配合物1—3的衍射数据采集于配备了CMOS PHOTON 100探测器与Mo Kα射线源(λ=0.710 73 Å,1 Å=0.1 nm)和Cu Kα射线源(λ=1.541 78 Å)的Bruker D8-Venture X射线单晶衍射仪上,所有晶体结构均采用直接法求解,由Olex2软件解析并采用F2的全矩阵最小二乘法(SHELXL-2017)进行结构精修。除氢原子采用各向同性热参数外,其它非氢原子均采用各向异性热参数修正。红外光谱的测量在Bruker Tensor 27红外光谱仪上进行。用光谱级的KBr稀释样品,压成片状。测量波数为400~4 000 cm-1。粉末X射线衍射数据采集于配备了Cu Kα射线源(λ=1.540 6 Å)的Bruker D8 Advance衍射仪上,测试范围是5°~50°(步长:0.02°)。配合物1和2的热重分析(TGA)由TA Q500分析仪记录,温度范围为30~800 ℃,在空气气氛中,升温速率为5 ℃/min。荧光光谱用F-4500型荧光分光光度计测量,配有氙灯和固体样品夹。光电倍增管电压为700 V,激发缝宽度为5.0 nm,发射缝宽度为5.0 nm,扫描速率为60 nm/min。用电子顺磁共振谱仪记录室温下的电子顺磁共振谱(EPR),由紫外可见分光光度计测量紫外-可见光谱。
1.2 晶体材料合成与表征
注意:硝酸铀酰原料中的铀主要为238U和极微量的235U(质量分数低于0.711%),属于放射性同位素,当使用铀酰化合物进行合成反应和表征时,应遵循处理放射性物质的标准防护措施。所有的化学试剂均为商业供应和使用的,没有进行进一步纯化。将UO2(NO3)2·6H2O(12.55 g, 0.025 mol)溶解在去离子水(50 mL)中,得到硝酸铀酰(0.5 mol/L)原液。有机配体1-(4-羧基苯基)-4,4′-联吡啶氯化物(Hhpcl·Cl)根据相关文献[30-32]报道进行合成。
本工作中所有的铀酰化合物均使用10 mL聚四氟乙烯衬里的Parr型热压釜进行水热合成。相应的合成条件记录如下。
(UO2)(PA)(hpcl)(配合物1):称取Hhpcl·Cl配体17.6 mg(0.05 mmol),邻苯二甲酸配体(H2PA)16.6 mg(0.1 mmol),加入到10 mL聚四氟乙烯反应釜中,并加入2 mL去离子水作为溶剂,再加入100 μL 0.5 mol/L硝酸铀酰溶液;充分混合后,将反应釜密封后升温至120 ℃,反应时间2 d;待反应釜冷却至室温后,过滤,用去离子水洗涤,得到黄色透明片状晶体;产率约为22%(基于铀)。
(UO2)(IPA)(hpcl)(配合物2):称取Hhpcl·Cl配体17.6 mg(0.05 mmol),间苯二甲酸配体(H2IPA)16.6 mg(0.1 mmol),加入到10 mL聚四氟乙烯反应釜中,并加入2 mL去离子水作为溶剂,再加入100 μL 0.5 mol/L硝酸铀酰溶液;充分混合后,将反应釜密封后升温至120 ℃,反应时间2 d;待反应釜冷却至室温后,过滤,用去离子水洗涤,得到黄色透明块状晶体;产率约为28%(基于铀)。
(UO2)(TPA)(hpcl)·(H2O)3·(H2TPA)0.5(配合物3):称取Hhpcl·Cl配体17.6 mg(0.05 mmol),对苯二甲酸配体(H2PTA)16.6 mg(0.1 mmol),加入到10 mL聚四氟乙烯反应釜中,并加入2 mL去离子水作为溶剂;再加入100 μL 0.5 mol/L硝酸铀酰溶液,最后加入100 μL 1 mol/L氢氧化钠溶液调节反应体系pH;充分混合后,将反应釜密封后升温至120 ℃,反应时间2 d;待反应釜冷却至室温后,过滤,用去离子水洗涤,得到少量黄色透明晶体,并伴随大量无定型沉淀。
2 结果与讨论
2.1 晶体结构解析
表1给出了配合物1—3的晶体学数据。

表1 配合物1—3的晶体学数据Table 1 Crystallographic data of compounds 1-3
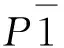

(a)——配合物1的不对称单元结构,(b)——辅助配体PA2-的配位模式,(c)——铀酰中心的协调领域,(d)——配合物1的一维链状结构图1 配合物1的晶体结构图Fig.1 Crystal structure of compound 1

(a)——配合物2的不对称单元结构,(b)——辅助配体IPA2-的配位模式,(c)——铀酰中心的协调领域,(d)——配合物2的一维链状结构图2 配合物2的晶体结构图Fig.2 Crystal structure of compound 2

(a)——配合物3的不对称单元结构,(b)——配合物3的Z型一维链状结构,(c)——六边形孔结构,(d)——二位超分子网络结构图3 配合物3的晶体结构图Fig.3 Crystal structure of compound 3

2.2 配合物1和2的表征
配合物1和2的X射线粉末衍射(PXRD)数据示于图4(a、b)。由图4(a、b)可以看出,其中黑色谱线代表两个单晶结构数据的模拟PXRD数据,红色线为合成样品的实验PXRD数据,模拟数据与实验数据的衍射峰位置吻合,证明所合成的两个结构均是纯相。

(a、b)分图中黑色谱线代表两个单晶结构数据的模拟PXRD数据,红色线为合成样品的实验PXRD数据图4 配合物1(a)和配合物2(b)的X射线粉末衍射数据、热重分析数据(c)及荧光性能(d)Fig.4 PXRD data of compound 1(a) and compound 2(b), and thermogravimetric results(c) and fluorescence characterization(d) of compounds 1 and 2
配合物1和2的TGA分析示于图4(c)。如图4(c)所示,两个配位聚合物均只有一个失重过程。其中配合物1的质量损失从380 ℃开始,到约450 ℃时分解完全;配合物2的质量损失从435 ℃开始,到约480 ℃时分解完全。虽然两个配合物的结构不同,但是元素组成完全相同,因此失重比例也相近,约为60%,与理论值60.5%吻合。这说明二者均具有良好的热稳定性能。
在相同的实验条件下研究了配合物1和2的室温固态发光性质,结果示于图4(d)。如图4(d)所示,在420 nm激发时,可以观察到配合物1和2中铀酰离子的特征荧光发射。在光谱中可以观察到配合物1的5个发射峰(492、513、536、560、588 nm),它们是铀酰阳离子的代表性荧光光谱峰。配合物1的最强峰位于513 nm。相比于UO2(NO3)·6H2O的发光,配合物1的荧光光谱轻微红移4 nm,但是荧光强度显著增强。这是因为含具有芳香环状共轭体系的配体在光照下产生了n-π*跃迁和π-π*跃迁,并且与铀酰生成配合物后,形成了更大的平面共轭体系,因此荧光强度增加。这一结果与文献[33-34]中报道的铀酰发射位移与赤道配体给电子能力之间的相关性一致。配合物2的荧光光谱与配合物1相似,发射峰的位置蓝移1 nm,荧光强度与配合物1相比略微下降。
2.3 配合物2的光致/热致变色性质研究
与其他基于紫精的化合物类似,配合物2具有明显的光致变色性质(图5)。如图5(a)所示,在紫外灯照射(365 nm、24 W)2 h后,配合物2的粉末样品从棕黄色缓慢变化为绿色。在室温下的黑暗环境中放置两周或在150 ℃下加热4 h后,这种绿色的样品又可以恢复到原来的颜色。为了进一步研究配合物2的光致变色行为,表征了其在不同时间紫外光照射下的紫外-可见光谱。如图5(b)所示,随着照射时间的增加,配合物2的紫外光谱在600~800 nm出现一个新的吸收峰,并在照射150 min后,此吸收峰强度达到最大值。对比相关文献[35]可知,这个新的吸收峰的出现是由于配合物2在紫外光照射下生成了紫精自由基。电子顺磁共振(EPR)光谱也进一步证实了这一结论。如图5(c)所示,配合物2的EPR光谱在g因子为2.005的位置存在一个尖锐的对称信号,并且随着紫外光照射而显著增强,说明紫外光照射对紫精自由基的产生有促进作用,同时,也导致材料荧光发射性能的减弱(图5(d))。

(a)——配合物2的光致变色和热致变色行为,(b)——紫外灯照射下配合物2的时间依赖性紫外可见光谱,(c)——室温下配合物2在未照射和紫外灯照射30 min的EPR光谱,(d)——紫外光照对配合物2的荧光发射影响图5 配合物2的光致变色和热致变色行为Fig.5 Photochromic and thermochromic behavior of compound 2
3 结 论
本课题组通过水热法合成了3例基于紫精配体的铀酰配位聚合物,并探索了它们的光化学性质,得到了如下结果。
3种配位聚合物的主体骨架均是一维链状结构,但是铀酰离子的配位模式完全不同。在配合物1中,相邻的2个铀酰离子通过2个邻苯二甲酸桥联,并扩展为一维链状结构;在配合物2中,相邻的2个铀酰离子通过2个间苯二甲酸分子桥联形成1个(UO2)2(IPA)2双核铀酰单元,这种双核单元再通过间苯二甲酸连接在一起,形成一维链状结构;而在配合物3中,对苯二甲酸配体不仅参与配合物一维链骨架的构筑,游离的对苯二甲酸分子还通过羧基O与紫精配体的吡啶N之间的氢键作用将一维链状结构扩展为二维超分子网络结构。
不同的辅助配体导致铀酰阳离子配位模式的不同,造成配位聚合物三维结构的多样化,最终导致材料性能的差异。例如有机配体的引入,显著增强了铀酰离子的荧光性能,配合物1和2在常温下均表现出明显的荧光发射行为。配合物2表现出了可逆的光致/热致变色性质。紫外-可见光谱和EPR光谱表明这种可逆的变色行为是由于配合物2在紫外光照射下、通过电子转移产生了紫精自由基所致。因此,辅助配体的引入可以有效提高铀酰配位聚合物的结构多样性,功能配体则可以赋予铀酰配合物独特的性能。
本工作对于新型锕系功能材料的设计、合成与应用研究具有一定的指导意义。
