pH-dependent Synthesis of Octa-nuclear Uranyl-oxalate Network Mediated by U-shaped Linkers
2020-03-08WUSiMEILeiHUKongQiuCHAIZhiFangNIEChangMingSHIWeiQun
WU Si, MEI Lei, HU Kong-Qiu, CHAI Zhi-Fang,3, NIE Chang-Ming, SHI Wei-Qun
pH-dependent Synthesis of Octa-nuclear Uranyl-oxalate Network Mediated by U-shaped Linkers
WU Si1,2, MEI Lei2, HU Kong-Qiu2, CHAI Zhi-Fang2,3, NIE Chang-Ming1, SHI Wei-Qun2
(1. School of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China; 2. Laboratory of Nuclear Energy Chemistry, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China; 3. Engineering Laboratory of Advanced Energy Materials, Ningbo Institute of Industrial Technology, Chinese Academy of Sciences, Ningbo 315201, China)
In this work, we report a novel octa-nuclear uranyl (U8) motif [(UO2)8O4(μ3-OH)2(μ2-OH)2]4+embedded in a uranyl-oxalate coordination polymer (compound 1) based on a U-shaped linker with extra-long xylylene chains for stabilizing the resulting high-nuclear motif through additional cross-linking connectivity. A comparison with dimeric and monomeric uranyl compoundsobtained at different pH value from the same hydrothermal system reveals that, solution pH plays a vital role in formation of this octa-nuclear uranyl motif by promoting hydrolysis of uranyl source. Since high similarity of eight uranium centers in this nearly planar U8 motif here, overlapping and broadening of signals in fluorescence, infra-red (IR) and Raman spectra can be found.
actinide coordination polymers; octa-nuclear uranyl; U-shaped Linker; pH effect
Because of the great importance in nuclear fuel cycle and environmental decontamination, actinide chemistry has gained increasing attentions from chemists, geologists and materials specialists[1-2]. As a naturally occurring actinide element, uranium, which is mostly featured by the form of hexavalent uranyl cation (UO22+) in ambient atmosphere, has been utilized as metal node in many uranium-based metal-organic compounds[3-6]. In close relation with the strong hydrolysis tendency of uranyl cation at varying aqueous pH values[7-9], a large variety of uranyl building units with different uranium nuclearities and/or connecting modes could be found in the large library of uranyl compounds[6]. Therefore, structural identification of uranyl compounds with diversified uranyl compositions in solid state can provide an alternative route to explore the solution chemistry of uranyl cation in terms of hydrolysis and speciation under different conditions. Most importantly, the well-defined structural and chemical features of these uranyl compounds with high-nuclear units could be helpful to get more insight on precise control of actinide hydrolysis and effective isolation of certain high-nuclear species from the actinide aqueous solution.
While many oligomeric uranyl units as well as infinite chain-like polynuclear uranyl motifs have been characterized in uranyl-organic compounds, few cases of uranyl-organic compounds with oligomeric octa-nuclear uranyl motifs[10-11]have been reported so far, though there are several octa-nuclear uranyl cage species constructed through coordination-driven assembly[12-13]. The scarcity of higher nuclearity might be attributed to harsh forming conditions for them as well as their instability in molecular structure.
Herein, a uranyl-oxalate coordination polymer containing a new type of oligomeric octa-nuclear uranyl motif, [(UO2)8O4-(3-OH)2(2-OH)2]4+, was isolated successfully and identified with the aid of a flexible U-shaped dicarboxylate linker (Xyl-BPy4CA) with an extra-long xylylene chain.
1 Experimental
1.1 Synthesis
(UO2)8O4(3-OH)2(2-OH)2(C2O4)2(C20H16N2O4)2(1) UO2(NO3)2·6H2O (200 μL, 0.10 mmol), [-Xyl- BPy4CEt]Br2(28.0 mg, 0.05 mmol), NaOH (3.2 mg, 0.08 mmol), ultrapure water (1.0 mL) was loaded into a 15 mL autoclave (the initial pH is 2.72), sealed and heated to 150 ℃ in an oven for 2 d, then automatically cooled to ambient temperature. Dark yellow crystals of 1 (Fig. S1) were produced, filtered off, and rinsed with ultrapure water and subjected to air-drying at room temperature to give 1 in pure phase in spite of preferred alignment of some crystal indices (Fig. S2). Yield: 14 mg (35% based on uranium).
[(UO2)2(2-OH)2(C20H16N2O4)2(H2O)2](NO3)2(2) compound 2 was synthesized using the same protocol except a reducing amount of NaOH (2.4 mg, 0.06 mmol). Prism yellow-green crystals were filtered off, and rinsed with ultrapure water and subjected to air-drying at room temperature to give 2 in pure phase (Fig. S1 and S3). Yield: 22 mg (31% based on uranium).
(UO2)(C2O4)(C6H5NO2) (3) compound 3 was synthesized using the same protocol like 1 and 2 but without NaOH added. Stick-like yellow-green crystals of 3 can be obtained as accompanied by considerable amount of 2 and other unidentified impurities (Fig. S1 and S4).
1.2 Characterization
1H-NMR spectra were recorded on a Bruker AVANCE III (500 MHz, Bruker, Switzerland) with deuterium oxide (D2O) as solvent. ESI-MS spectra were obtained with a Bruker AmaZon SL ion trap mass spectrometer (Bruker, USA). Powder X-ray diffraction (PXRD) measurements were recorded on a Bruker D8 Advance diffractometer with Cu Kα radiation (=0.15406 nm) in the range 5°–60° (step size: 0.02°). Thermogravimetric analysis (TGA) was performed on a TA Q500 analyzer over the temperature range of 25–800 ℃ in air with a heating rate of 10 ℃/min. Solid-state fluorescence spectra were measured on a Hitachi F-4600 fluorescence spectrophotometer. The Fourier transform infrared (IR) spectra were recorded from KBr pellets in the range of 4000–400 cm–1at Bruker Tensor 27 spectrometer. Raman spectra were recorded on a Thermo Fisher Scientific DXRxi Micro Raman imaging spectrometer excited at 780 nm.
Uranyl-oxalate coordination polymer (namely compound 1, Fig. 1(a)) containing a new type of oligomeric octa- nuclear uranyl motif, [(UO2)8O4-(3-OH)2(2-OH)2]4+(Fig. 1(b)), was isolated successfully and identified with the aid of a flexible U-shaped dicarboxylate linker (Xyl-BPy4CA) with an extra-long xylylene chain (Fig. 1(c-d)). By a direct comparison with another uranyl compounds (namely compound 2 and compound 3) with monomeric or dimeric uranyl nodes from the same organic ligand, factors affecting the formation of compound 1 such as aqueous pH and hydrothermal stability of organic ligand have been discussed. The physicochemical properties of compound 1 were also characterized and compared in detail.
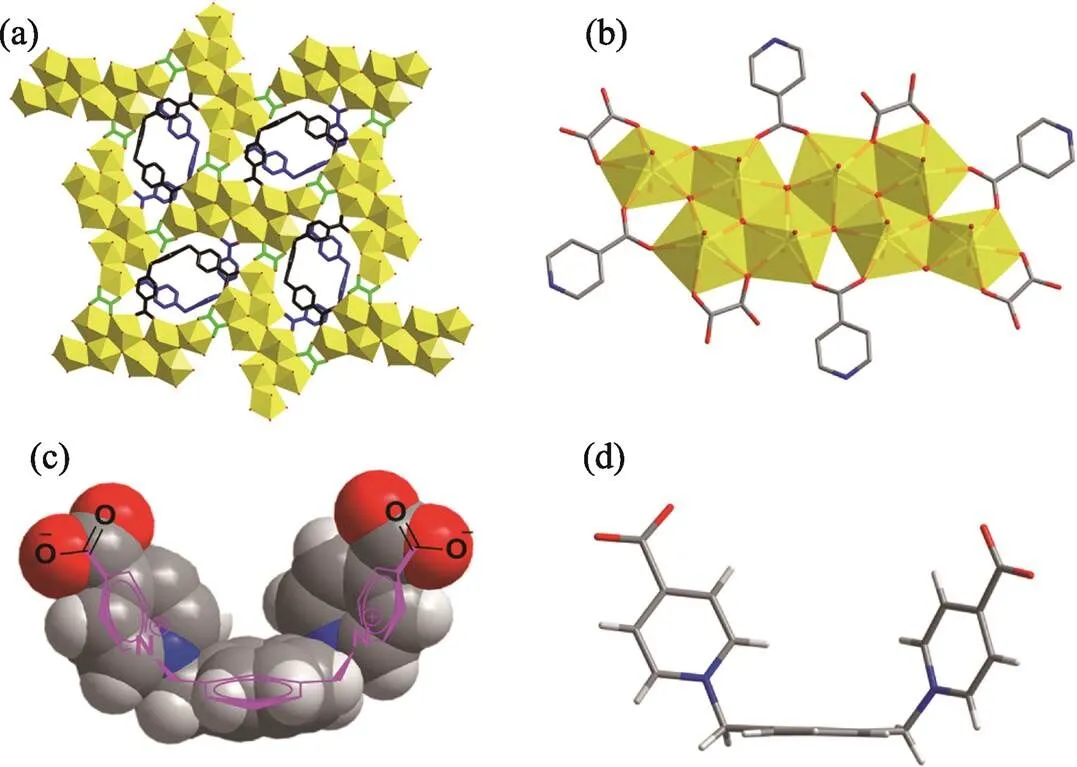
Fig. 1 Octa-nuclear uranyl-oxalate network reinforced by U-shaped zwitterionic dicarboxylate linkers
(a) two-dimensional coordination network; (b) octa-nuclear uranyl (U8) motif, [(UO2)8O4(3-OH)2(2-OH)2]4+; (c) U-shaped linker in a space- filling mode overlapped with its molecular structure; (d) U-shaped linker in a stick mode
Color codes: uranyl polyhedra in yellow; U-shaped linkers in dark or blue
2 Results and discussion
2.1 Structural description
Crystallographic analysis shows that compound 1 crystallizes in the monoclinic crystal system with a space group of P21/c, and its asymmetric unit contains four unique uranium centers, a linker of-Xyl-BPy4CA ([C20H16N2O4]) and an-formed oxalate ligand ([C2O4]2–) (Fig. 2(a)). The formation of oxalate ion is more or less a little surprising in spite of several similar cases found in other heterocycle-based or labile organic ligands[13].Viewing from the extending structure of the asymmetric unit, an elongated U8 motif can be found (Fig. 2(b)), among which every uranium center is in a pentagonal bipyramid (P-type) geometry with equatorial U–O bond distances in the range of 0.2193(14)–0.2578(14) nm (Table S1), and links to each other through sharing equatorial edges with other neighboring polyhedrons.Based on the bond valence sum analysis[14], the bond valence value of O(11) is 1.393, which should be assigned to μ3-hydroxo group, while the atoms of O(9) and O(10) are μ3-oxo groups (bond valence values of 1.943 and 2.20, respectively), and O(12) is μ2-hydroxo group (bond valence values of 1.137). The feature of μ3-hydroxo group for O(11)is ascribed to weak binding affinity to U(3) and U(4). Compared with the cation-cation interactions (CCIs) directed U8 motif reported previously[11], the [(UO2)8O4(3-OH)2(2-OH)2]4–motif found here represents a new type of uranyl oligomer with a nearly planar geometry (Fig. S5).
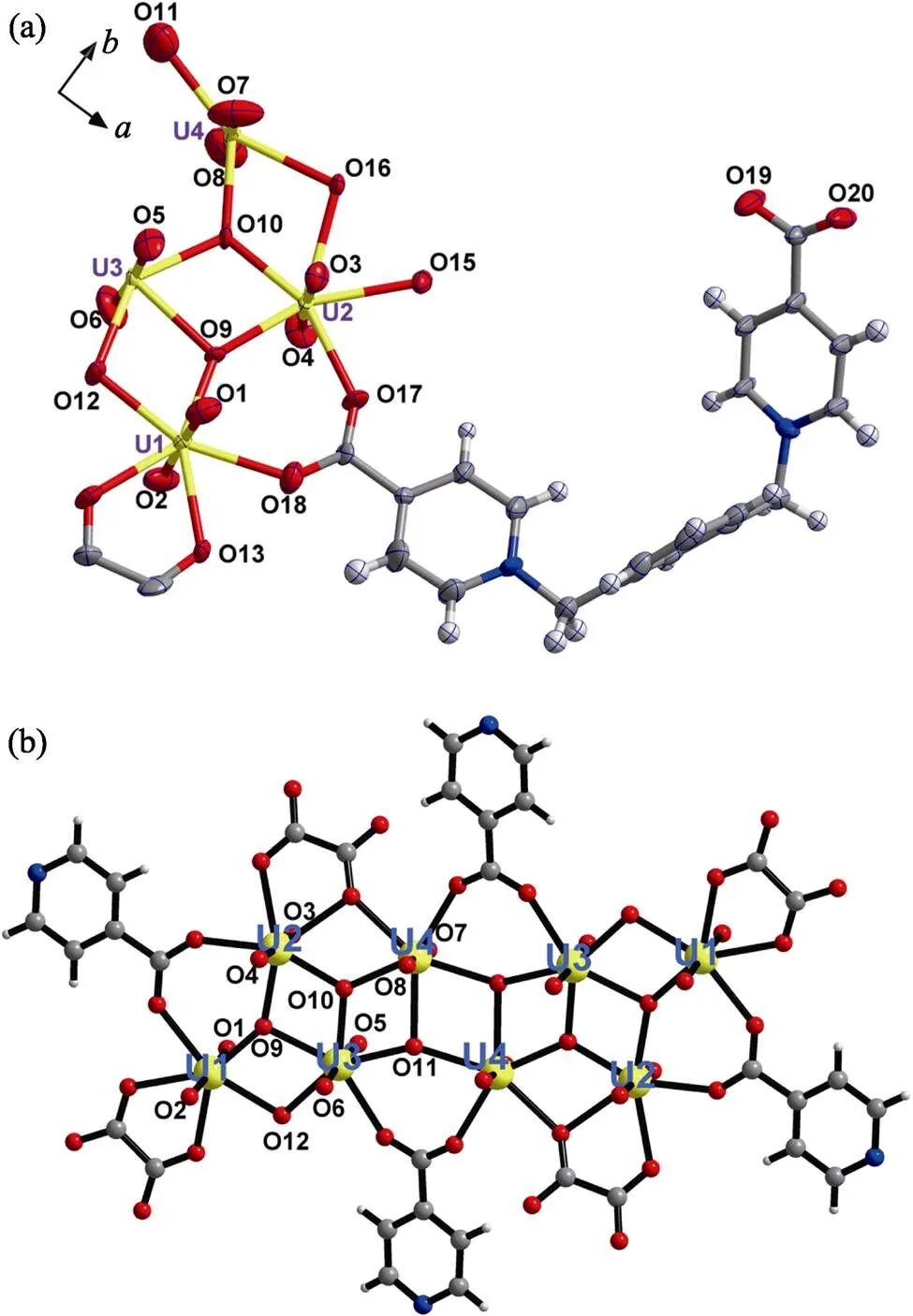
Fig. 2 Crystal structure of compound 1
(a) ORTEP view of compound 1 with the 30% probability level for thermal ellipsoids; (b) octa-nuclear uranyl (U8) motif in compound 1 showing detailed coordination spheres of all uranyl centers
Color codes: uranium atoms in yellow; oxygen atoms in red; carbon atoms in dark gray; nitrogen atoms in blue; hydrogen atoms pale gray
Each U8 motif is eight-connected with four oxalate and four-Xyl-BPy4CA moieties surrounded (Fig S6(a)). The oxalate ligands, always go together with a U-shaped bidentate-Xyl-BPy4CA, promote the extension of U8 motif from four directions through connecting four adjacent ones in a bridging mode (one side is in2-2:1mode and the other side is in2mode) (Fig. S6(b)), and subsequently lead to a 2D network with the minimum rhombic loop in size of 1.193 nm×1.077 nm (Fig. S6(c-d)). Detailed analysis reveals that, every U8 motif displays a different overall orientation from that of its adjacent U8 with an angle of inclination of 36.6(4)°, which is contributed to the distortion of the above rhombic loop (Fig. S7).
When the amount of sodium hydroxide added to the hydrothermal system of uranyl and [-Xyl-BPy4CEt]Br2decreased gradually, another two uranyl compounds 2 and 3 emerge. Compound 2 is a molecular compound with a dimeric uranyl motif containing two equivalent uranyl centers (Fig. 3(a-b)). Each uranyl is in a pentagonal bipyramid geometry with equatorial U–O bond distances in the range of 0.2325(2)–0.2576(2) nm (Table S1). Besides two bridging hydroxyl groups and two water molecules, the uranyl dimer is coordinated by two bidentate1-mode-Xyl-BPy4CA linkers both in a head-to-tail way, and finally gives a pattern of twinned double loops (Fig.3(b)). The simple double loops interact with two nitrate anions by hydrogen bonds (Fig. S8and Table S2) and stack with each other to form the final crystal lattice of molecular compound (Fig. 3(c-d)).
Another uranyl compound 3 with a one-dimensional (1D) chain-like topology was produced under the more acidic condition. As the case of compound 1, the-formed oxalate ligand ([C2O4]2–) is present in compound 3, indicating the frequent occurrence of oxalate ion for the hydrothermal systems of pyridine derivatives[13]such as [-Xyl-BPy4CEt]Br2used here. In the crystal structure of compound 3, not only oxalate ligands that bridge monomeric uranyl groups to form a one-dimensional chain, but also protonated isonicotinate as a ligand at both sides can be found (Fig. 4). The presence of protonated isonicotinate in compound 3 suggests the higher degrees of decomposition of [-Xyl-BPy4CEt]Br2under more acidic condition compared with that found in compound 1 and 2.
2.2 pH-dependent regulation on the formation of U8 motif
The distinct structural difference among compounds 1, 2 and 3 suggests the significant impact of pH on the uranyl hydrothermal systems, especially the uranyl speciation and crystallization process (Fig. 5). The strong dependence on pH of uranyl speciation is largely attributed to hydrolysis of uranyl ion in aqueous solution. Generally speaking, uranyl monomer is likely to be stable at low pH, whereas high pH of aqueous solution always induces uranyl hydrolysis and promotes its oligomerization through the olation or oxolation process. With the aid of certain organic ligands, these oligomeric uranyl species could further transfer to the solid state as poly-nuclear uranyl compounds. Resembling most uranyl systems in aqueous solution, the formation of U8 motif in compound 1 also follows a pH-regulated mechanism. As revealed by the synthesis of compound 1–3, the aqueous pH used for compound 1 (pH ~2.72) with octa-nuclear uranyl motifs is higher than that for compounds 2 (pH ~2.29) and 3 (pH ~1.35) with only dimeric or monomeric uranyl motifs. Interestingly, pH also exerts a non-negligible influence on the stabilization of organic ligands under hydrothermal conditions. In contrast to the large degree of decomposition of [-Xyl-BPy4CEt]Br2to simple isonicotinate linkers found in compound 3, the original zwitterionic dicarboxylate linkers still retains its main molecular skeleton with controlled hydrolysis to its acid form of-Xyl-BPy4CA during hydrothermal reaction with uranyl under less acidic conditions for the synthesis of compound 1. It can be speculated that the preservation of total skeleton of-Xyl-BPy4CA with strong coordination capability toward uranyl units as demonstrated in compound 1 also contributes to the formation of U8 motifs by affording additional stabilization through coordination and cross- linkage. The role of U-shaped-Xyl-BPy4CA linkers in the stabilization of high-nuclear U8 motifs will be discussed as followed.
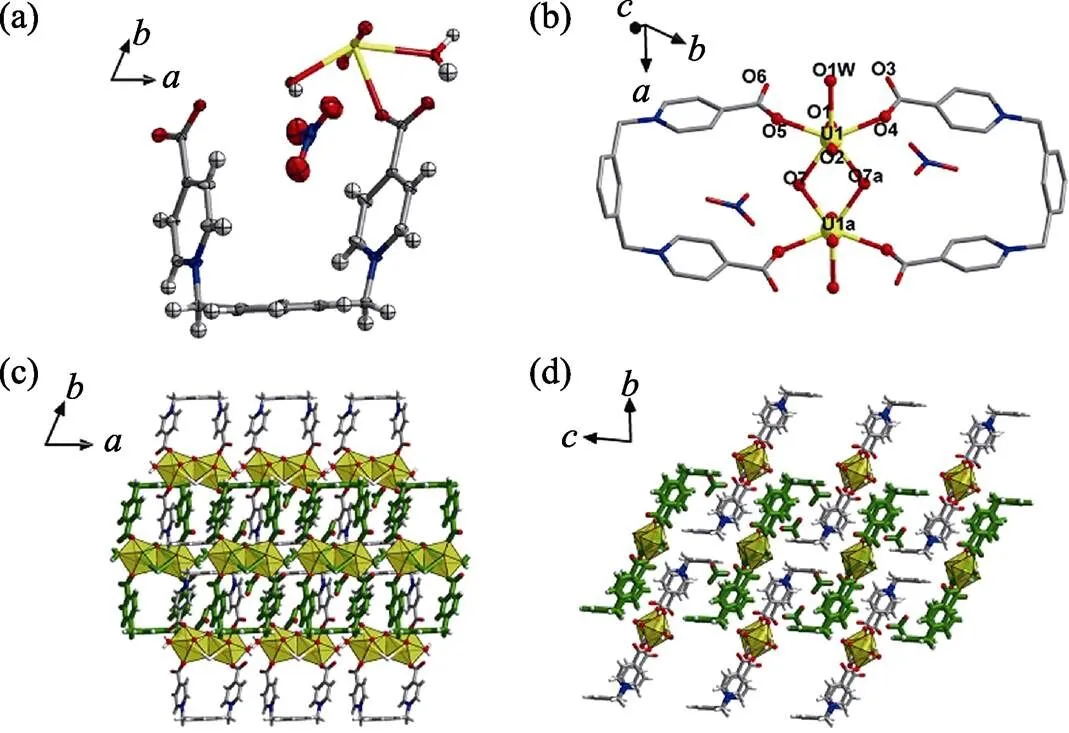
Fig. 3 Crystal structure of compound 2
(a) ORTEP view of compound 2 with the 30% probability level for thermal ellipsoids; (b) coordination environment of each uranyl center for dimeric uranyl motif; (c-d) crystal lattice stacking for compound 2 viewed foraxis (c) andaxis (d)
Color codes: uranium atoms in yellow; oxygen atoms in red; carbon atoms in dark gray; nitrogen atoms in blue; hydrogen atoms pale gray; the U-shaped linkers in green
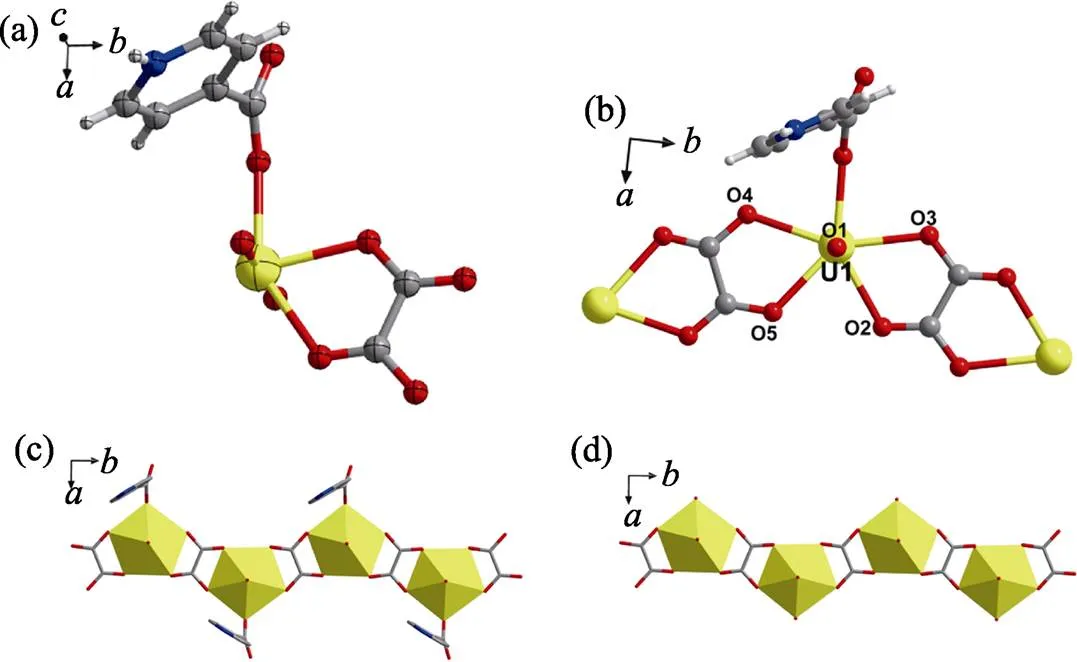
Fig. 4 Crystal structure of compound 3
(a) ORTEP view of compound 3 with the 30% probability level for thermal ellipsoids; (b) coordination environments of uranyl center; (c-d) the extended structure based on one-dimensional oxalate-bridging monomeric uranyl chain with (c) or without (d) terminal isonicotinate ligands
Color codes: uranium atoms or polyhedras in yellow; oxygen atoms in red; carbon atoms in dark gray; nitrogen atoms in blue; hydrogen atoms in pale gray
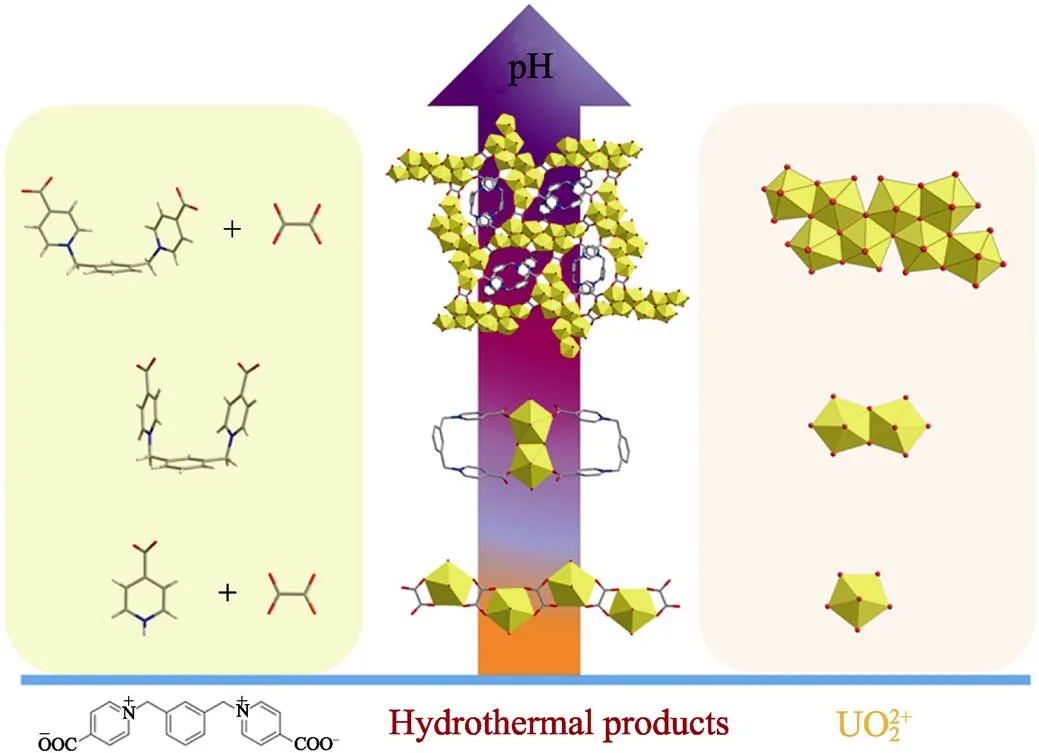
Fig. 5 pH-dependent regulation of hydrothermal reactions of m-Xyl-BPy4CA linkers and uranyl
Color codes: uranium polyhedras in yellow; oxygen atoms in red; carbon atoms in gray; nitrogen atoms in blue
2.3 Structural stabilization of high-nuclear U8 motifs
It is interesting to find that most of uranyl-oxalate compounds[14-20]only afford uranyl secondary building units with nuclearities of no more than 4. The difference of compound 1 from those reported previously in uranyl oligomerization suggests that there are other additional factors contributing to the stabilization of U8 motif with more uranyl units. Apparently, structural stabilization based on coordination bonds and/or chelating effects of organic ligands is one of the vital factors to promote the final crystallization of U8-bearing product as companied with the pH-induced formation of U8 motifs. TheXyl-BPy4CA linker plays dual roles in the construction of U8-based compound 1, including realizing coordination saturation of uranium centers of U8 and additional structural connectivity for rhombic loop as well as crystal packing. Specifically, two bidentateXyl-BPy4CA ligands in a couple crosslink all the four U8 motifs through coordination bonds and hydrogen bonds, where oneXyl-BPy4CA ligand points upwards and the other points downwards from the opposite direction (Fig.S9). The coordination bonds in a bridgingμ-η1:1mode on both ends ofXyl-BPy4CA joint two adjacent U8 motifs through two binding patterns (lateral and terminal binding sites), while hydrogen bonds between the C-H groups on the ‘back’ of U-shaped linker and oxygen atoms from another two U8 motifs of the rhombic loop consolidate the location ofXyl-BPy4CA in the cavity. Moreover, the important role ofXyl-BPy4CA also reflects on its participation of hydrogen bonds between adjacent layers of 2D sheets through interacting with neighboured uranyl group from another sheet, which mainly contributes to the crystal packing of compound 1 in three-dimensional (3D) space (Fig. S10). In all, the participation ofXyl-BPy4CA linker increases the molecular connectivity, enhances the structural stability of high-nuclear metal clusters and thus promotes the formation of new high-nuclear uranyl motifs.
Moreover, a comprehensive survey on the relationship between high-nuclear uranyl motifs and organic ligands used reveals that, multi-topic organic ligand with a nonlinear configuration is more likely to fit for capture, fixation and stabilization of high-nuclear uranyl motifs by coordination to uranyl units in a compact way (Fig. S11). Another important feature ofXyl-BPy4CA linker in molecular structure is its flexible conformation (Fig. S12). This U-shaped ligand could display varying molecular conformations in different compounds so as to be adapted to different coordination environment of uranyl motif. For example, theXyl-BPy4CA linker gives a more open conformation when located at the cavity of uranyl-oxalate network in compound 1 (angle between two pyridium groups is ~47.9°, left in Fig. S12), while it becomes more compact with two dangled pyridium groups nearly paralleled to each other (angle between two pyridium groups is ~13.8°, right in Fig. S12). As shown above, the flexibility of molecular skeleton forXyl-BPy4CA linker ensures it structural adaptivity and facilitates the formation of new U8 motif.
2.4 Physicochemical Properties
Characterizations of properties including thermogravimetric analysis (TGA), Fourier transform infrared (IR) spectra, Raman spectra, and solid-state fluorescence spectra for compounds 1 and 2 in pure phase were conducted. In terms of thermal stability, compound 1 does not undergo thermal decomposition of oxalate group on the backbone of 2D network until the temperature increases up to ~295 ℃ (Fig. S13), which is in sharp contrast to compound 2 (Fig. S14). The good stability of compound 1 is similar to the thermal behavior of uranyl oxalate[21]. IR spectrum of compound 1 (Fig. S15) shows only a weak broad signal at ~915 nm correspondding to the characteristic vibrations of axial U=O bonds (calculated data based on the empirical equation of [d (U=O) (pm)=10650[1(cm–1)]–2/3+57.5][22]: 0.175–0.179 nm), although there are eight uranium centers in the secondary building unit of compound 1. The asymmetric stretching vibration of axial U=O bonds for compounds 2 and 3 (911 and 910 nm, respectively) is comparable to that observed for compound 1 (Fig. S15). The Raman spectrum of compound 1 (Fig. S16) also displays only two peaks assigned to asymmetric ν3vibrations (calculated data based on the empirical equation of [d (U=O) (pm) = 9141 [3(cm–1)]–2/3+ 80.4][22]: 0.175–0.178 nm), which is very similar to that observed for compound 3 with monomeric uranyl unit. A comparison of compound 1 with previously-reported CCI- mediated U8 motif[11]in IR and Raman spectra shows that the latter one gives finer signals of stretching vibrations that could be resolved well and assigned to different uranyl centers among the U8 motif, while the IR and Raman signals for different uranyl centers of U8 motif in compound 1 seem to be highly overlapped with each other. This phenomenon might be, by contrasted to CCI-based U8 motif, ascribed to the nearly planar attribute of non-CCI U8 motif, which partly offsets the differences between uranyl centers of U8 motif in compound 1. In short, difference in physicochemical properties between this type of U8 motif and that mediated by CCIs reflects the particularity of U8 found here.
As shown in Fig. S17, solid-state fluorescence spectra of compounds 1 and 2 are more or less different as compared to that of uranyl nitrate (UO2(NO3)2). Compound 2 gives the typical vibronic progression of uranium (VI) with the five main emission bands located at 499, 520, 543, 568 and 596 nm corresponding to the S11→ S00and S10→ S0ν(ν=0−4) electronic transitions[23]. It is very similar to that for a uranyl terephthalate compound with the same dimeric uranyl units[24], and shows a large red-shift (7−11 nm) compared to that of uranyl nitrate. The large red-shift observed here should be attributed to markedly different coordination geometries and coordination modes for uranyl centers in compound 2 and UO2(NO3)2[25-28]. Interestingly, the fluorescence of compound 1 gives an unresolved broad signal with a broad peak ranging from 530 to 550 nm, and also turns to own a red-shift as well as a little attenuation in intensity compared to those of compound 2 and uranyl nitrate. Resembling the situation of IR and Raman spectra, the peak broadening might be attributed to the signal overlapping of uranium(VI) centers in the nearly planar U8 motif of compound 1[25]. Considering the complexity for figuring out complicated interactions between different uranyl units in this large octa-nuclear uranyl cluster, the precise assignment and analysis of fluorescence spectra is still difficult here, and further study on this issue with the aid of modeling methods might be needed in the near future.
3 Conclusions
In summary, we have successfully synthesized a uranyl- oxalate coordination polymer with a new type of octa- nuclear uranyl motif, [(UO2)8O4-(μ3-OH)2(μ2-OH)2]4+by utilizing U-shaped pyridinium-based organic ligand [-Xyl-BPy4CEt]Br2a pH-dependent regulation. The mixed ligand system of oxalateandXyl-BPy4CA formedplays important roles for stabilizing high-nuclear uranyl species by bridging coordination linkage and superimposed structural connectivity, respectively. The U8-bearing uranyl compound found here provides more information on uranyl hydrolysis and speciation in aqueous solution, and also facilitates the exploration of new actinide materials by employing well- designed organic linkers.
Supporting Materials:
Supporting Materials related to this article can be found at https://doi.org/10.15541/jim20190118
[1] ALTMAIER M, GAONA X, FANGHANEL T,. Recent advances in aqueous actinide chemistry and thermodynamics., 2013, 113(2): 901–943.
[2] JONES M B, GAUNT A J. Recent developments in synthesis and structural chemistry of nonaqueous actinide complexes., 2013, 113(2): 1137–1198.
[3] WANG K X, CHEN J S. Extended structures and physicochemical properties of uranyl-organic compounds., 2011, 44(7): 531–540.
[4] ANDREWS M B, CAHILL C L. Uranyl bearing hybrid materials: synthesis, speciation, and solid-state structures., 2013, 113(2): 1121–1136.
[5] YANG W T, PARKER T G, SUN Z M,. Structural chemistry of uranium phosphonates., 2015, 303(1): 86–109.
[6] LOISEAU T, MIHALCEA I, HENRY N,. The crystal chemistry of uranium carboxylates., 2014, 266(35): 69–109.
[7] RAI D, FELMY A R, RYAN J L,. Uranium (IV) hydrolysis constants and solubility product of UO2·H2O(am)., 1990, 29(2): 260–264.
[8] AHRLAND S. On the complex chemistry of the uranyl ion Ι. The hydrolysis of the 6-valent uranium in aqueous solutions., 1949, 3(4): 374–400.
[9] ZANONATO P, DI BERNARDO P, BISMONDO A,. Hydrolysis of uranium (VI) at variable temperatures (10–85 ℃)., 2004, 126(17): 5515–5522.
[10] SALMON L, THUERY P, EPHRITIKHINE M,. Crystal structure of the first octanuclear uranium (IV) complex with compartmental schiff base ligands., 2004, 23(4): 623–627.
[11] MIHALCEA I, HENRY N, CLAVIER N,. Occurence of an octanuclear motif of uranyl isophthalate with cation-cation interactions through edge-sharing connection mode., 2011, 50(13): 6243–6249.
[12] PASQUALE S, SATTIN S, ESCUDERO-ADAN E C,. Giant regular polyhedra from calixarene carboxylates and uranyl., 2012, 3(1): 785.
[13] THUERY P. A highly adjustable coordination system: nanotubular and molecular cage species in uranyl ion complexes with kemp's triacid., 2014, 14(3): 901–904.
[14] WANG L H, SHANG R, ZHENG Z,. Two systems of [DabcoH2]2+/[PipH2]2+-uranyl-oxalate showing reversible crystal-to- crystal transformations controlled by the diammonium/uranyl/ oxalate ratios in aqueous solutions ([DabcoH2]2+=1,4-diazabicyclo- [2.2.2]-octaneH2and [PipH2]2+= PiperazineH2)., 2013, 13(6): 2597–2606.
[15] CHAPELET-ARAB B, NOWOGROCKI G, ABRAHAM E,. Crystal structure of new uranyl oxalates (NH4)2[UO2(C2O4)·2H2O] and (NH4)2–x(N2H5)[UO2(C2O4)3]·3H2O (=0 and=1). Comparison with other uranyl oxalates., 2005, 93(5): 279–285.
[16] GIESTING P A, PORTER N J, BURNS P C,. A series of sheet-structured alkali metal uranyl oxalate hydrates: structures and IR spectra., 2006, 221(8): 589–599.
[17] GIESTING P A, PORTER N J, BURNS P C,. Uranyl oxalate hydrates: structures and IR spectra., 2006, 221(4): 252–259.
[18] DUVIEUBOURG L, NOWOGROCKI G, ABRAHAM F,. Hydrothermal synthesis and crystal structures of new uranyl oxalate hydroxides:- and-[(UO2)2(C2O4)(OH)2(H2O)2] and [(UO2)2((C2O4)(OH)2(H2O)2]·H2O., 2005, 178(11): 3437–3444.
[19] THUERY P. Reaction of uranyl nitrate with carboxylic diacids under hydrothermal conditions. Crystal structure of complexes with L(+)-tartaric and oxalic acids., 2007, 26(1): 101–106.
[20] VOLOGZHANINA A V, SEREZHKINA L B, NEKLYUDOVA N A,. Synthesis and characterisation of a trinuclear uranyl complex: crystal structure of (CN3H6)5[(UO2)3O(OH)2(CH3COO)(C2O4)3]., 2009, 362(14): 4921–4925.
[21] CHUGH C A, SHARMA A, SHARMA A,. Kinetics and mechanism of thermal decomposition of uranyl oxalate., 2011, 23(4): 1865–1866.
[22] BARTLETT J R, COONEY R P,. On the determination of uranium oxygen bond lengths in dioxouranium (VI) compounds by raman-spectroscopy., 1989, 193(1): 295–300.
[23] BRACHMANN A, GEIPEL G, BERNHARD G,. Study of uranyl (VI) malonate complexation by time resolved laser-induced fluorescence spectroscopy (TRLFS)., 2002, 90(3): 147–153.
[24] MEI L, WANG C Z, ZHU L Z,. Exploring new assembly modes of uranyl terephthalate: templated syntheses and structural regulation of a series of rare 2d→3d polycatenated frameworks., 2017, 56(14): 7694–7706.
[25] NATRAJAN L S. Developments in the photophysics and photochemistry of actinide ions and their coordination compounds., 2012, 256(15/16): 1583–1603.
[26] THUERY P, HARROWFIELD J. Solvent effects in solvo-hydrothermal synthesis of uranyl ion complexes with 1,3-adamantanediacetate., 2015, 17(21): 4006– 4018.
[27] THUERY P, HARROWFIELD J. Structural variations in the uranyl/4,4'-biphenyldicarboxylate system. rare examples of 2d→3d polycatenated uranyl-organic networks., 2015, 54(16): 8093–8102.
[28] THUERY P, RIVIERE E, HARROWFIELD J,. Uranyl and uranyl-3d block cation complexes with 1,3-adamantanedicarboxylate: crystal structures, luminescence, and magnetic properties., 2015, 54(6): 2838–2850.
pH调控合成U型配体介导的八核铀酰草酸网络
吴思1,2, 梅雷2, 胡孔球2, 柴之芳2,3, 聂长明1, 石伟群2
(1. 南华大学 化学化工学院, 衡阳 421001;2. 中国科学院 高能物理研究所 核能放射化学实验室, 北京 100049; 3. 中国科学院 宁波材料技术与工程研究所, 先进能源材料工程实验室, 宁波 315201)
本工作报道了一种含新型八核铀酰(U8)团簇单元([(UO2)8O4(μ3-OH)2(μ2-OH)2]4+)的草酸铀酰配合物, 该化合物中, U型有机配体链可以增强铀酰之间的交联度, 具有稳定多核铀酰团簇的作用。通过与另外两种含单核和双核的铀酰配位化合物比较, 发现八核铀酰团簇单元的形成是一个pH调控的过程。理化性质分析显示, 荧光、红外、拉曼的信号峰都出现了不同程度的重叠和宽化, 表明八个铀酰离子具有较高的相似度, 这与此多核铀酰团簇的近平面分子构型密切相关。
锕系配位聚合物;八核铀酰中心;U-型链;pH调控
Supporting materials:
pH-dependent Synthesis of Octa-nuclear Uranyl-Oxalate Network Mediated by U-shaped Linkers
WU Si1,2, MEI Lei2, HU Kong-Qiu2, CHAI Zhi-Fang2,3, NIE Chang-Ming1, SHI Wei-Qun2
(1. School of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China; 2. Laboratory of Nuclear Energy Chemistry, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China; 3. Engineering Laboratory of Nuclear Energy Materials, Ningbo Institute of Industrial Technology, Chinese Academy of Sciences, Ningbo 315201, China)
S1. General Methods
S1. 1 Synthesis
1,1¢-(1,3-phenylenebis(methylene))bis(4-(ethoxycarbonyl)pyridin-1-ium) bromide ([-Xyl-BPy4CEt]Br2).[-Xyl-BPy4CEt]Br2was synthesized according to the reported procedure[5-7].A mixture of 1, 3-Bis(bromomethyl)benzene (0.65 g, 2.46 mmol) and isonicotinate (0.83 g, 5.49 mmol) were dissolved in 50 mL of acetonitrile and refluxed for 48 h. After cooling to room temperature, the solution was concentrated by evaporation in vacuum, filtered, washed with acetone, and dried under vacuum to afford the final product.Yield: 1.287 g (92.8%).1H NMR (500 MHz, D2O,ppm): 9.11 (d, 4H); 8.54 (d, 4H), 7.59–4.63 (m, 4H), 5.95 (s, 4H), 4.51 (q, 4H), 1.42 (t, 6H). MS (ESI): mass calculated for C24H26N2O42+(M2+), 406.19; m/z found, 203.11 (M2+/2).
S1. 2 X-ray single crystal structure determination
X-ray diffraction data of compound 1 and 2 was performed on Bruker D8 VENTURE X-ray CMOS diffractometer with a Mo Kα X-ray source (=0.071073 nm) at 296 K. X-ray diffraction data of compound 3 was collected on a Agilent SuperNova X-ray CCD diffractometer with a Cu Kα X-ray source (=0.154184 nm) with higher diffraction capability at 293 K. Standard Agilent Crysalis software was used for the determination of the unit cells and data collection control. All the crystal structures were solved by means of direct methods and refined with full-matrix least squares on SHELXL- 2014. SIMU were used to constrain the displacement parameters of the phenyl and pyridyl groups and ISOR were used to even out the electron density associated with disordered portions of the moieties for both 1 and 3. OMIT were used to eliminate bad reflections obscured by the beamstop for all compounds. Since there is high disorder of pyridyl groups dangling aside, the pyridyl ligand was forced to be half occupied to create a chemically sensible model for 3. Solvent molecules (water) in the structure are highly disordered and impossible to be modelled as discrete atomic sites. To resolve this issue, the contribution of solvent-electron density was removed using the SQUEEZE/PLATON procedure, thereby producing a set of solvent-free diffraction intensities used for improving the structure refinements. The crystal data of both compounds are given in Table S3. Crystallographic data for the structures in this study were deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC- 1510791 (1), CCDC-1898268 (2) and CCDC-1510792 (3).
S2. Figures

Fig. S1 Different optical morphologies of 1 with octa-nuclear uranyl (U8) motifs, 2 with binuclear uranyl (U2) motifs and 3 with monomeric uranyl (U1) motifs
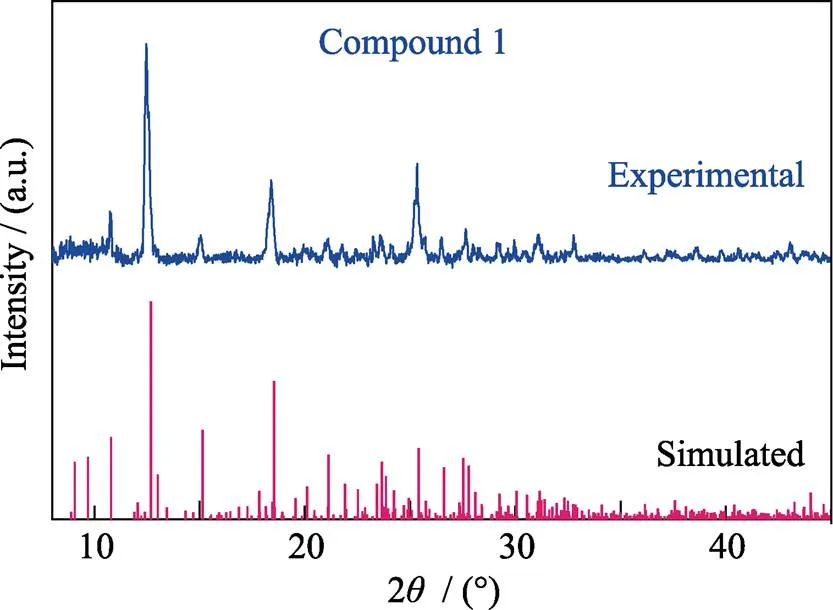
Fig. S2 Experimental and simulated patterns of powder X-ray diffraction (PXRD) of compound 1
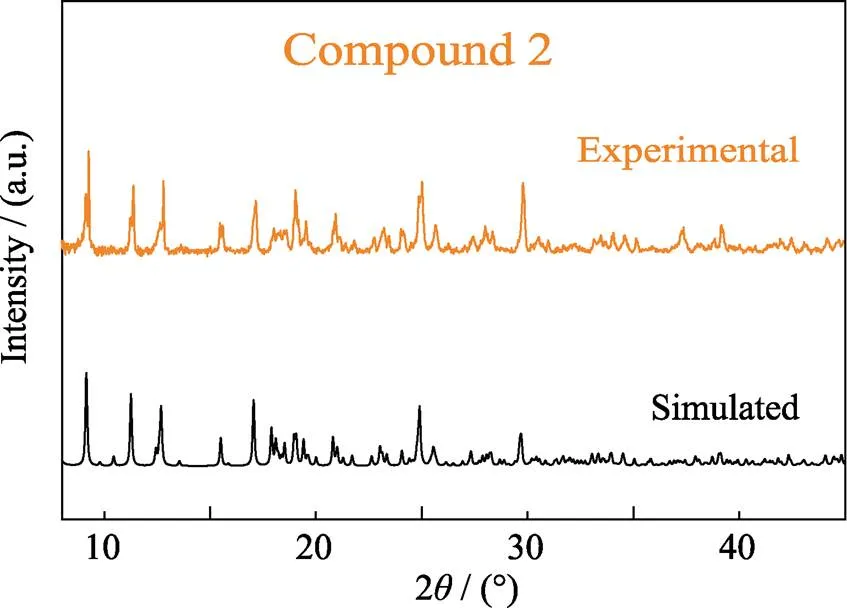
Fig. S3 Experimental and simulated patterns of powder X-ray diffraction (PXRD) of compound 2
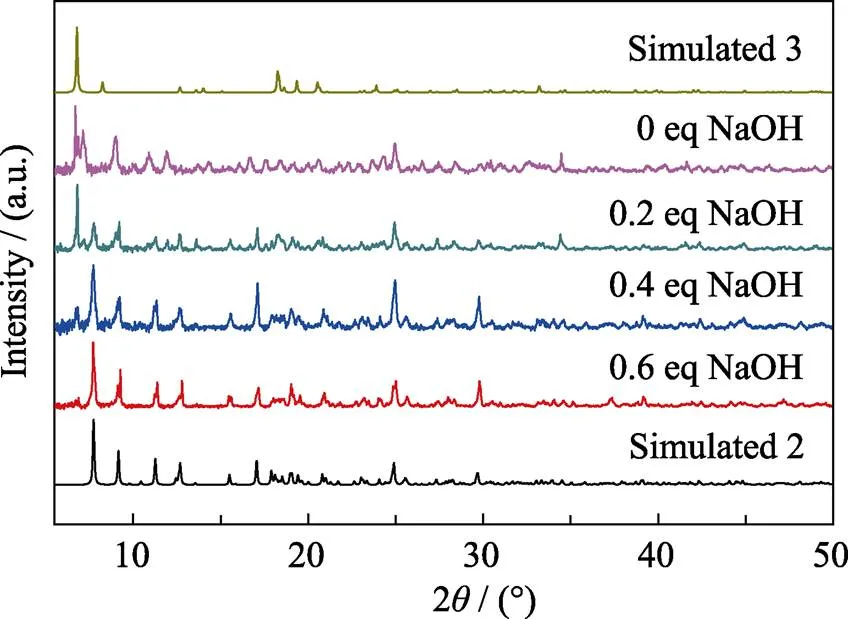
Fig. S4 Experimental and simulated patterns of powder X-ray diffraction (PXRD) of compound 3
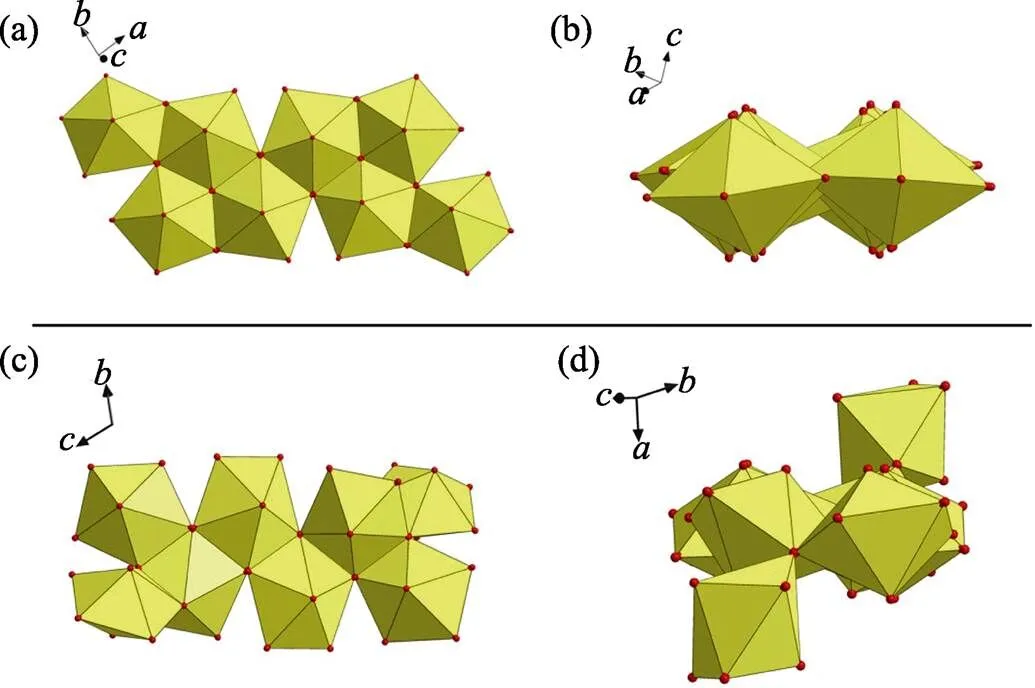
Fig. S5 (a) A nearly planar geometry of U8 motif found in this work; (b) a non-planar U8 motif with cation-cation interactions (CCIs) reported by Loiseau,[1]
Fig. S6 (a-b) Eight-connected U8 motif with four oxalate (Ox) and fourXyl-BPy4CA (L) moieties extends from four directions through oxalate ligands (a), which thus connecting four adjacent ones with each oxalate ligand going together with a U-shaped bidentateXyl-BPy4CA linker (b); (c) U8-based uranyl-oxalate 2D network (enlarged diagram: a minimum rhombic loop); (d) U8-based uranyl-oxalate 2D network with all the cross-linkingXyl-BPy4CA linkers omitted for clarity (enlarged diagram: a minimum rhombic loop in size of 1.193 nm× 1.077 nm)
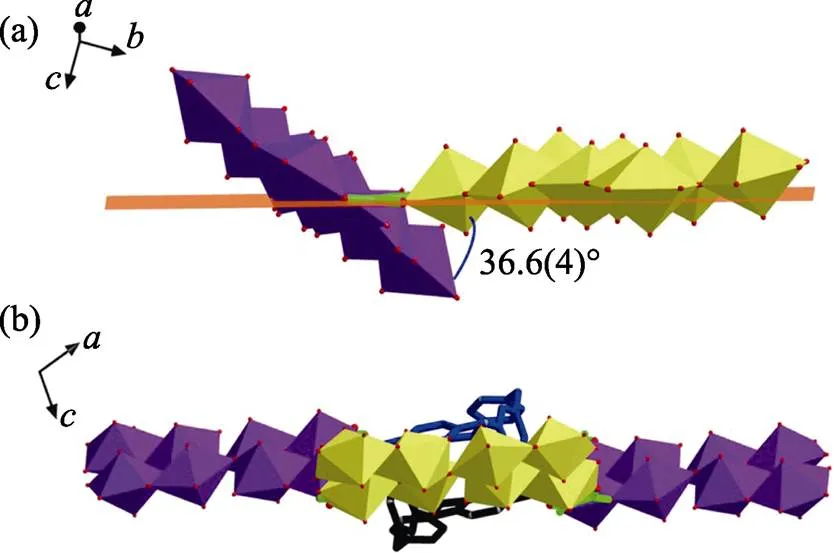
Fig. S7 Each U8 motif displays a different overall orientation from that of its adjacent U8 with an angle of inclination of 36.6(4)° (a), resulting in a distortion of the rhombic loop (b)
Fig. S8 Hydrogen bonds between double loops and two nitrate anions
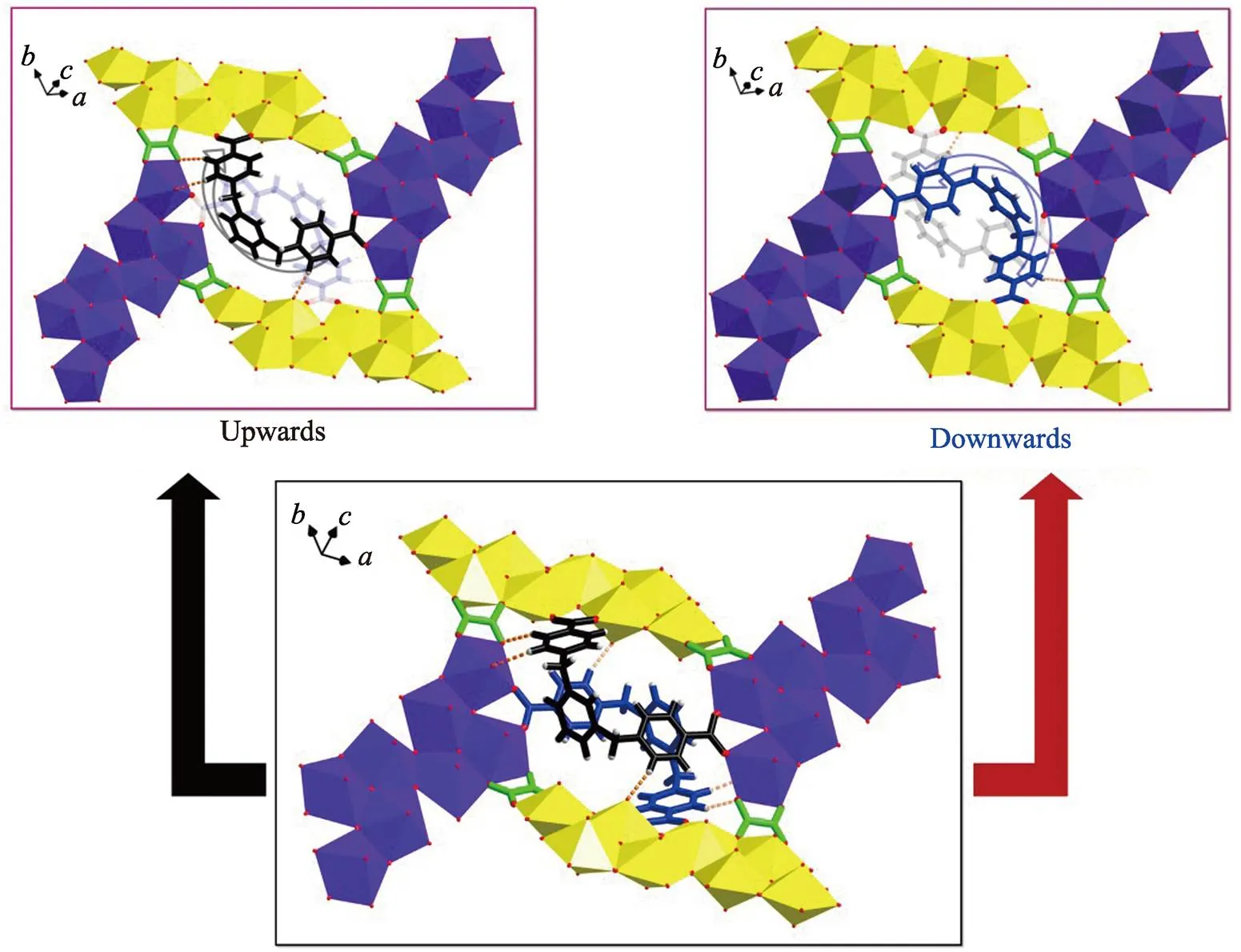
Fig. S9 Two ‘U’-shaped bidentate-Xyl-BPy4CA ligands located in the cavity of rhombic loop crosslink all the four U8 motifs through coordination bonds and hydrogen bonds (bottom) where one-Xyl-BPy4CA ligand points upwards (top left) and the other points downwards from the opposite direction (top right)
Fig. S10 Hydrogen bonds between adjacent layers of 2D sheets through U8 motifs that interact with neighbouredXyl-BPy4CA from another sheet orXyl-BPy4CA interacting with neighboured uranyl group from another sheet
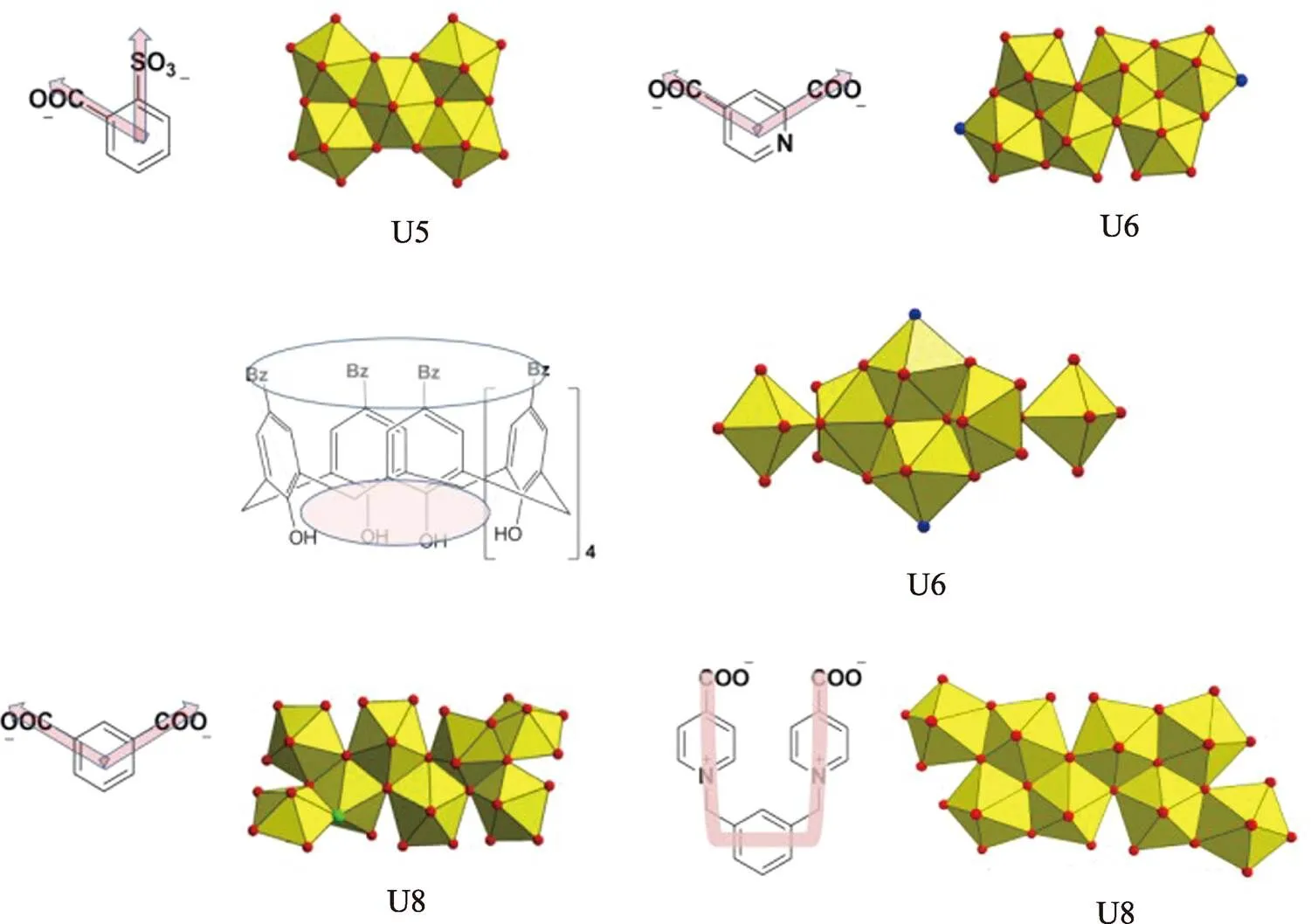
Fig. S11 Some examples of high-nuclear uranyl motif based on nonlinear multi-topic organic ligands, as suggested by the cases of pentanuclear (U5), hexanuclear (U6) and octanuclear (U8) uranyl motifs derived from sulfobenzoate precursors[2],-position or-position aromatic/heteroaromatic dicarboxylate[3-4],calixarene ligand[3]and U-shaped linkers used in this work
Fig. S12 Different molecular conformation of-Xyl-BPy4CA linker in 1 and 2 demonstrating its flexibility in molecular conformation
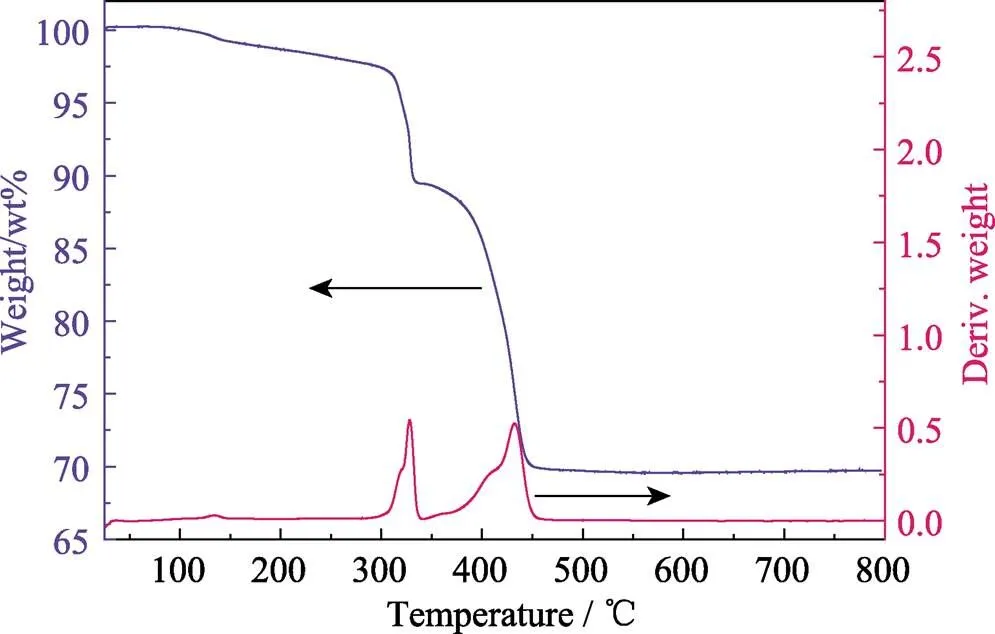
Fig. S13 Thermogravimetric analysis (TGA) of compounds 1, where 1 starts to decompose at ~295 ℃, and finally transforms to U3O8with residual weight of 69.31% (theoretical value: 70.25%)
Fig. S14 Thermogravimetric analysis (TGA) of compounds 2, where 2 starts to decompose at ~233 ℃, and finally transforms to U3O8with residual weight of 40.95% (theoretical value: 40.20%)
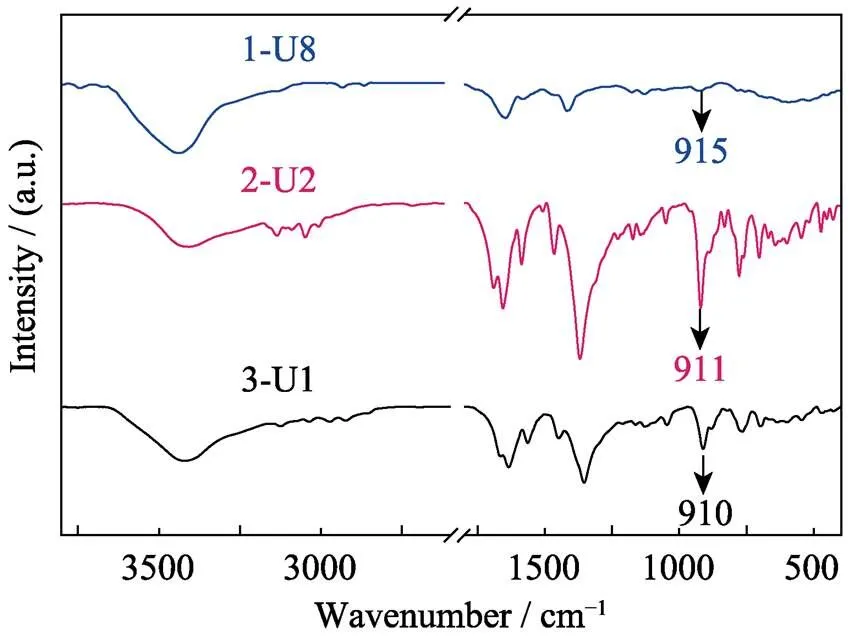
Fig. S15 Fourier transform infrared (IR) spectra of compounds 1 (U8 motif, blue line), 2 (U2 motif, red line) and 3 (U1 motif, black line) with characteristic symmetric1vibrations at 915, 911 and 910 nm, respectively
Fig. S16 The Raman spectra of compounds s 1 (U8 motif) and 3 (U1 motif) with characteristic asymmetric ν3 vibrations (1: 833 and 863 cm-1; 3: 829 and 860 cm-1)
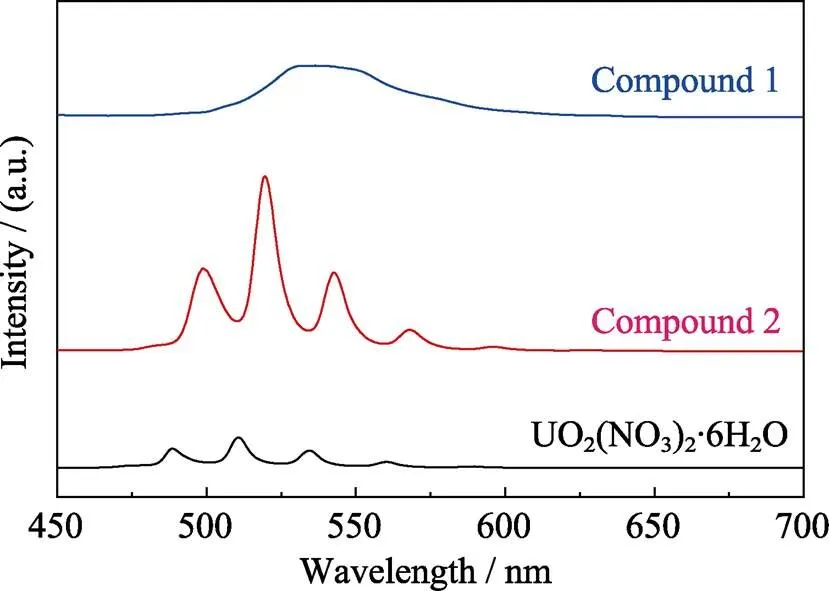
Fig. S17 Solid-state fluorescence spectra of compound 1 and 2 as compared to that of uranyl nitrate (UO2(NO3)2): 1, a broad peak ranging from 530 to 550 nm; 2, five main emission bands located at 499, 520, 543, 568 and 596 nm; UO2(NO3)2, 488, 511, 534, 561 and 589 nm
Fig. S181H NMR of [-Xyl-BPy4CEt]Br2(500 MHz, 298 K, D2O)
S3. Tables
Table S1 Selected bond distances related to uranyl centers in compounds 1, 2 and 3
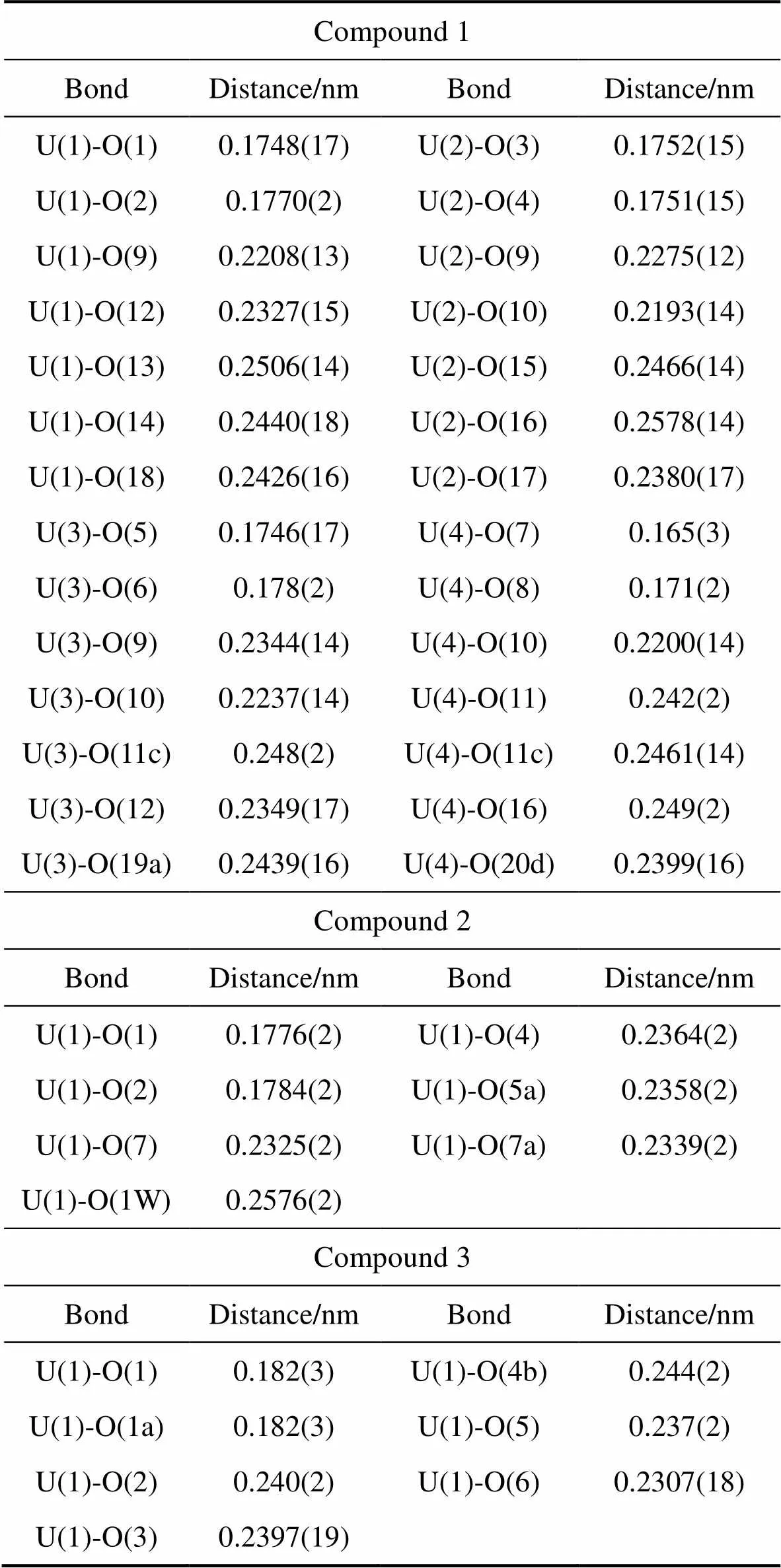
Compound 1 BondDistance/nmBondDistance/nm U(1)-O(1)0.1748(17)U(2)-O(3)0.1752(15) U(1)-O(2)0.1770(2)U(2)-O(4)0.1751(15) U(1)-O(9)0.2208(13)U(2)-O(9)0.2275(12) U(1)-O(12)0.2327(15)U(2)-O(10)0.2193(14) U(1)-O(13)0.2506(14)U(2)-O(15)0.2466(14) U(1)-O(14)0.2440(18)U(2)-O(16)0.2578(14) U(1)-O(18)0.2426(16)U(2)-O(17)0.2380(17) U(3)-O(5)0.1746(17)U(4)-O(7)0.165(3) U(3)-O(6)0.178(2)U(4)-O(8)0.171(2) U(3)-O(9)0.2344(14)U(4)-O(10)0.2200(14) U(3)-O(10)0.2237(14)U(4)-O(11)0.242(2) U(3)-O(11c)0.248(2)U(4)-O(11c)0.2461(14) U(3)-O(12)0.2349(17)U(4)-O(16)0.249(2) U(3)-O(19a)0.2439(16)U(4)-O(20d)0.2399(16) Compound 2 BondDistance/nmBondDistance/nm U(1)-O(1)0.1776(2)U(1)-O(4)0.2364(2) U(1)-O(2)0.1784(2)U(1)-O(5a)0.2358(2) U(1)-O(7)0.2325(2)U(1)-O(7a)0.2339(2) U(1)-O(1W)0.2576(2) Compound 3 BondDistance/nmBondDistance/nm U(1)-O(1)0.182(3)U(1)-O(4b)0.244(2) U(1)-O(1a)0.182(3)U(1)-O(5)0.237(2) U(1)-O(2)0.240(2)U(1)-O(6)0.2307(18) U(1)-O(3)0.2397(19)
Table S2 Distances and angles for hydrogen bonds observed in compounds 1 and 2
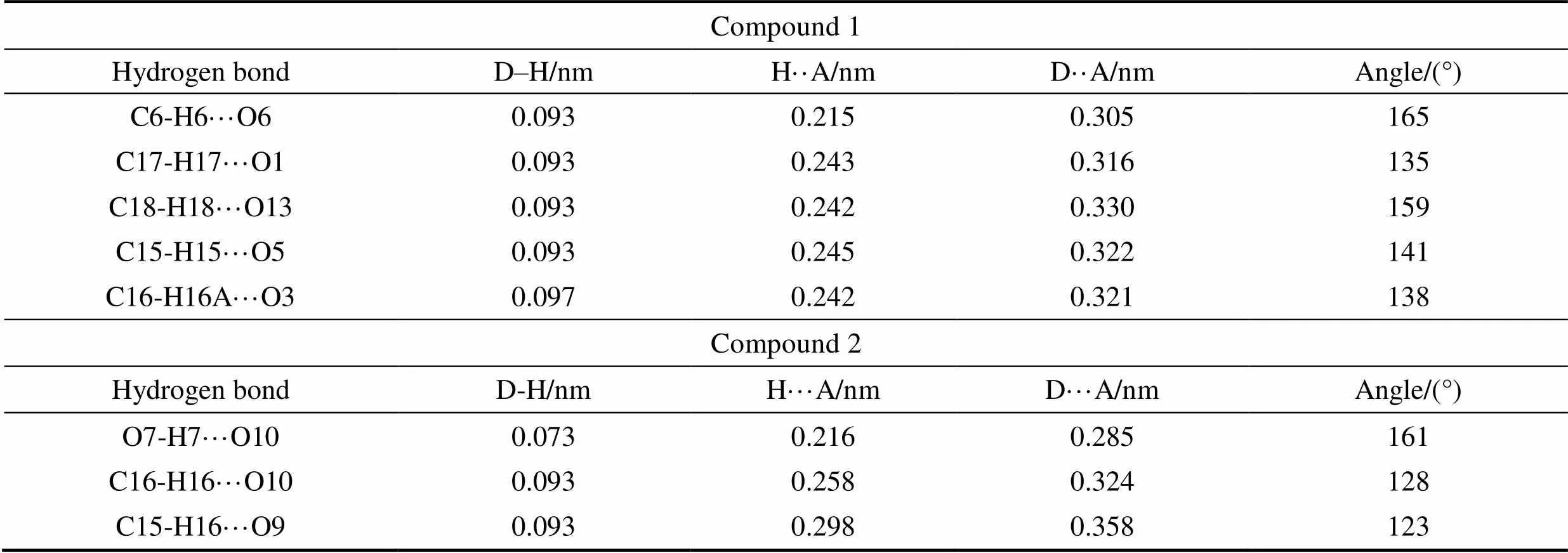
Compound 1 Hydrogen bondD–H/nmH··A/nmD··A/nmAngle/(°) C6-H6···O60.0930.2150.305165 C17-H17···O10.0930.2430.316135 C18-H18···O130.0930.2420.330159 C15-H15···O50.0930.2450.322141 C16-H16A···O30.0970.2420.321138 Compound2 Hydrogen bondD-H/nmH···A/nmD···A/nmAngle/(°) O7-H7···O100.0730.2160.285161 C16-H16···O100.0930.2580.324128 C15-H16···O90.0930.2980.358123
Table S3 Crystal data and structure refinement for compounds 1, 2 and 3
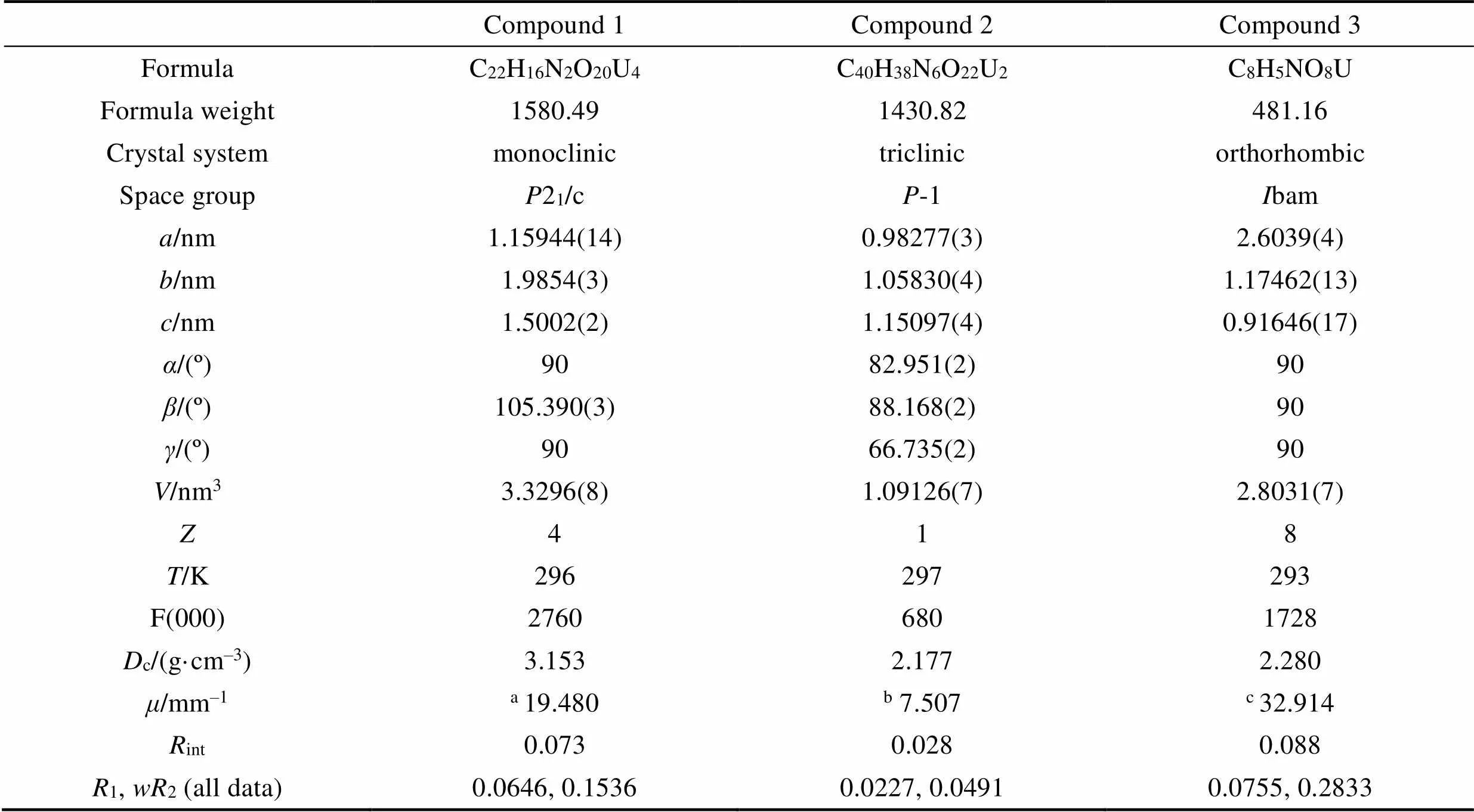
Compound 1Compound 2Compound 3 FormulaC22H16N2O20U4C40H38N6O22U2C8H5NO8U Formula weight1580.491430.82481.16 Crystal systemmonoclinictriclinicorthorhombic Space groupP21/cP-1Ibam a/nm1.15944(14)0.98277(3)2.6039(4) b/nm1.9854(3)1.05830(4)1.17462(13) c/nm1.5002(2)1.15097(4)0.91646(17) α/(º)9082.951(2)90 β/(º)105.390(3)88.168(2)90 γ/(º)9066.735(2)90 V/nm33.3296(8)1.09126(7)2.8031(7) Z418 T/K296297293 F(000)27606801728 Dc/(g·cm–3)3.1532.1772.280 μ/mm–1a 19.480b 7.507c32.914 Rint0.0730.0280.088 R1, wR2 (all data)0.0646, 0.15360.0227, 0.04910.0755, 0.2833
a, bMo K: 0.071073 nm;cCu K: 0.154184 nm
[1] MIHALCEA I, HENRY N, CLAVIER N,. Occurence of an octanuclear motif of uranyl isophthalate with cation–cation interactions through edge-sharing connection mode., 2011, 50(13): 6243–6249.
[2] THU RY P. Sulfonate complexes of actinide ions: structural diversity in uranyl complexes with 2-sulfobenzoate., 2013, 52(1): 435–447.
[3] ZHENG Y Z, TONG M L, CHEN X M,. Synthesis, structure and photoluminescent studies of two novel layered uranium coordination polymers constructed from UO (OH) polyhedra and pyridinedicarboxylates., 2005, (20): 4109–4117.
[4] THU RY P, NIERLICH M, SOULEY B,. Complexation of a hexameric uranium (VI) cluster by p-benzylcalix [7] arene., 1999, (15): 2589– 2594.
[5] SINDELAR V, MOON K, KAIFER A E,. Binding selectivity of cucurbit[7]uril: bis(pyridinium)-1,4-xylylene versus 4,4¢- bipyridinium guest sites., 2004, 6(16): 2665– 2668.
[6] HUANG F, SLEBODNICK C, MAHAN E J,. [3]Pseudorotaxanes based on the cryptand/monopyridinium salt recognition motif., 2007, 63(13): 2875–2881.
[7] MEI L, WANG L, YUAN L Y,. Supramolecular inclusion- based molecular integral rigidity: a feasible strategy for controlling the structural connectivity of uranyl polyrotaxane networks., 2015, 51(60): 11990–11993.
TQ174
A
1000-324X(2020)02-0243-07
10.15541/jim20190118
2019-03-21;
2019-04-27
National Natural Science Foundation of China (21671191, 21577144, 11405186)
WU Si (1993–), female, Master candidate. E-mail: wusi@ihep.ac.cn
吴思(1993–), 女, 硕士研究生. E-mail: wusi@ihep.ac.cn
SHI Wei-Qun, professor. E-mail: shiwq@ihep.ac.cn; NIE Chang-Ming, professor. E-mail: niecm196132@163.com
石伟群, 教授. E-mail: shiwq@ihep.ac.cn; 聂长明, 教授. E-mail: niecm196132@163.com
