ERK5抑制剂XMD17-109通过下调ITPRIP表达抑制胶质瘤进展*
2023-12-26王昕雯曹长春孙增先
王昕雯,曹长春,朱 亮,杜 宇,孙增先
(1.徐州医科大学附属连云港医院药学部,江苏连云港 222061;2.徐州医科大学淮安临床学院药学部,江苏淮安 223300;3.南京医科大学附属淮安第一医院药学部,江苏淮安 223300)
胶质瘤(glioblastoma,GBM)被认为是恶性程度最高、治疗难度最大的原发性脑肿瘤之一,发病率占恶性脑肿瘤的45%以上[1-3]。尽管多学科管理和靶向治疗在临床方面取得了一定进展,但恶性胶质瘤的现有治疗方案仍然较为局限[4-5]。抑癌基因的失活是胶质瘤不断进展的重要原因,也是治疗的主要障碍之一[6-8]。因此,深入了解这一过程所涉及的分子机制对于开发更有效的干预措施至关重要。细胞外信号调节激酶5(extracellular signal-regulated kinase 5,ERK5)是丝裂原活化蛋白激酶家族中的一员,具有独特的C末端结构域[9-10]。越来越多的证据表明ERK5参与肿瘤起始和进展的各个阶段[11-13]。研究认为,ERK5信号级联的激活会诱导多种细胞周期分子增殖[14]。此外,ERK5对周期蛋白依赖性激酶抑制因子的表达具有下调作用[15]。药物干预和基因策略明确证实,靶向ERK5通路对于癌症治疗具有潜力[16-17]。基因表达谱分析发现ERK5 mRNA在原发性脑肿瘤,特别是GBM中明显高表达[18]。新型小分子激酶抑制剂XMD17-109作为ERK5强效抑制剂被成功合成。XMD17-109在生化上抑制ERK5,IC50为(0.162±0.006)μmol/L,在细胞内阻断表皮生长因子诱导的ERK5自磷酸化。目前XMD17-109用于胶质瘤治疗的研究尚未见报道,且XMD17-109作用机制有待进一步探究。1,4,5-三磷酸受体(inositol 1,4,5-trisphosphate receptor,IP3R)相互作用蛋白已被证明是IP3R的结合伴侣,其与IP3R相互作用促进IP3R通道的钙离子抑制功能。前期研究已经证明肌醇1,4,5-三磷酸受体相互作用蛋白(inositol 1,4,5-trisphosphate receptor interacting protein,ITPRIP)通过连接肌球蛋白调节轻多肽9和死亡相关蛋白激酶1促进GBM进展[19]。本研究假设ERK5在胶质瘤中可能作为肿瘤促进ITPRIP的刺激分子。通过研究ERK5抑制剂XMD17-109对胶质瘤细胞U251活力的影响及ERK5与ITPRIP之间的相互作用,可揭示ERK5介导促进ITPRIP表达的分子机制,及其在胶质瘤进展中的意义,现报道如下。
1 材料与方法
1.1 材料
1.1.1细胞
本研究中人胶质母细胞瘤细胞株U251来自中国科学院分子细胞科学卓越创新中心。
1.1.2试剂
XMD17-109购于美国MCE公司;DMEM高糖培养基和胎牛血清(FBS)购于美国Gibco公司;Lipofectamine 2000转染试剂购于美国Thermo Fisher公司;PrimeScriptTMRT Master Mix和SYBR &Premix Ex TaqTM试剂盒购于日本TaKaRa公司;BCA试剂盒和十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)试剂购于上海碧云天生物技术有限公司;ERK5抗体购于美国Cell Signaling公司(1∶1 000,#12950);ITPRIP抗体(1∶1 000,26055-1-AP)和GAPDH抗体(1∶5 000,60004-1-Ig)购于美国Proteintech公司;辣根过氧化物酶(HRP)-山羊抗小鼠IgG抗体(1∶5 000,PA2201)和HRP-山羊抗兔IgG抗体(1∶5 000,PA2202)购于南京普诺恩生物技术有限公司;CCK8检测试剂盒购于南京建成公司;7-氨基放线菌素D(7-AAD)/藻红蛋白(PE)试剂盒和Transwell小室购于美国BD公司;ERK5、阴性对照siRNA(siNC)序列和质粒由上海GenePharma公司设计并合成。
1.1.3仪器
Applied biosystemsTM7500荧光定量PCR仪、酶标仪、培养箱和超净台购于美国Thermo Fisher公司;流式细胞仪购于美国Beckman公司;高速冷冻离心机购于芬兰Eppendorf公司;Amersham Biosciences蛋白成像系统购于美国Cytiva公司。
1.2 方法
1.2.1细胞培养
细胞在含有10% FBS的DMEM高糖培养基中,37 ℃、5% CO2环境常规培养。在体外细胞实验中,使用浓度为1 μmol/L的XMD17-109处理细胞24 h以抑制ERK5活性(XMD17-109组),未处理的细胞纳入Control组。
1.2.2RNA干扰
按照说明书提示方法,分别将ERK5 siRNA(siERK5)和siNC使用Lipofectamine 2000进行细胞转染,构建ERK5敲低和阴性对照模型,分为siERK5组、siNC组。转染6 h后,更换为含10% FBS的新鲜培养基。细胞连续培养48 h后,收集细胞进行蛋白、RNA分离等研究。通过逆转录实时荧光定量PCR(RT-qPCR)测定ERK5敲低效率。siERK5组和siNC组所对应RNA序列分别为5′-CGU GCC CUA UGG CGA AUU CAA TT-3′和5′-UUC UCC GAA CGU GUC ACG UTT-3′。
1.2.3质粒转染
人ERK5基因经过扩增和逆转录合成互补DNA(cDNA)。随后,将人ERK5 cDNA克隆pCDH-CMV-MCS-EF1-copGFP-T2A-Puro载体中。将含有ERK5和对照结构的溶液引入到U251细胞中,孵育48 h,构建ERK5过表达和对照结构模型,分别分为ERK5-OE组、Vector组。采用RT-qPCR和Western blot检测证实ERK5在细胞中的过表达水平。进一步将ERK5-OE组、Vector组细胞分别与XMD17-109进行孵育,分为ERK5-OE+XMD17-109组、Vector+XMD17-109组。
1.2.4总RNA提取及RT-qPCR
转染siRNA后,用TRIzol试剂提取细胞总RNA。使用纳米微滴设备评估总RNA的纯度和浓度。使用PrimeScriptTMRT Master Mix进行RNA逆转录,然后通过SYBR &Premix Ex TaqTM进行RT-qPCR检测。采用Ct(2-ΔΔCt)法(ΔΔCt=ΔCttarget-ΔCtGAPDH)对所有靶基因进行标准化,以GAPDH为内参基因。引物序列见表1。

表1 RT-qPCR引物序列
1.2.5Western blot
细胞收集后在冰上冷却30 min,使用含有20 mmol/L Tris、1 mmol/L EDT A、150 mmol/L氯化钠、1 mmol/L乙二醇双(2-氨基乙基醚)四乙酸(EGTA)、1%脱氧胆酸盐、1% Triton X-100,2 mmol/L正钒酸钠的裂解缓冲液进行蛋白质提取。随后,裂解液在4 ℃下以13 000 r/min离心20 min。所得上清液使用BCA试剂盒测定。将每个样品中等量的蛋白质与SDS样品缓冲液相结合,通过SDS-PAGE分离蛋白质,并将其转移到聚偏二氟乙烯(PVDF)膜上。蛋白转移完毕后,用含有5%脱脂奶粉的TBST(TBS中含0.1%吐温20)封闭1 h。然后将膜与ERK5抗体、ITPRIP抗体和GAPDH抗体在4 ℃下过夜孵育。用TBST清洗3次,每次清洗5 min。加入二抗,室温下孵育1 h,其中ERK5和ITPRIP选用HRP-山羊抗兔IgG二抗,GAPDH选用HRP-山羊抗小鼠IgG二抗。用增强化学发光检测系统对膜进行检测。使用Image J软件进行吸光度进行分析,每个蛋白条带的信号强度基于相对于各自的GAPDH加载对照进行归一化。
1.2.6CCK-8和细胞增殖实验
使用CCK-8检测试剂盒评估细胞增殖能力。在96孔板中每孔加入100 μL密度为3×104/mL的细胞混悬液。在5% CO2的培养箱中孵育0、24、48、72 h。随后,每孔加入10 μL的CCK-8试剂,继续孵育4 h后用酶标仪测定450 nm处吸光度(A)值,每组条件均设置3个复孔。
1.2.7流式细胞仪检测细胞凋亡
采用7-AAD/PE法检测U251细胞凋亡情况。将U251细胞与7-AAD/PE在冰上孵育15 min,检测细胞凋亡情况,应用Flowjo软件进行定量分析。
1.2.8划痕实验和Transwell实验
在划痕实验中,将细胞以2×106个/孔的密度重悬于6孔板中。一旦细胞贴壁,使用20 μL移液器吸头在细胞表面做出划痕。每个实验组均设置3个复孔。分别于划痕后0、24 h在显微镜下观察并拍照。每个孔中测量3个随机位置,使用Image J软件量化划痕宽度。按照说明书提示方法使用Transwell小室进行细胞迁移和侵袭实验。将1×105个细胞接种于上室500 μL无血清培养基中,下室加入750 μL含10% FBS的新鲜培养基,将细胞在37 ℃下孵育36 h。然后将迁移过滤网的细胞用4%多聚甲醛(PFA)固定,并用结晶紫溶液染色20 min。用棉签去除残留在上室表面的细胞后,在显微镜下随机拍照5个视野,采用直接计数法用Image J软件对迁移和侵袭细胞数量进行计数,取平均值作为最终迁移或侵袭细胞数量。
1.3 统计学处理
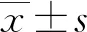
2 结 果
2.1 XMD17-109对ERK5和ITPRIP的影响
Control组与XMD17-109组ERK5 mRNA和蛋白表达水平无明显变化;与Control组相比,XMD17-109组ITPRIP mRNA和蛋白表达水平明显降低(P<0.05);与Control组相比,XMD17-109组吸光度降低,U251细胞增殖、侵袭和迁移能力均明显降低,细胞凋亡率明显升高,差异有统计学意义(P<0.05),见图1、2。
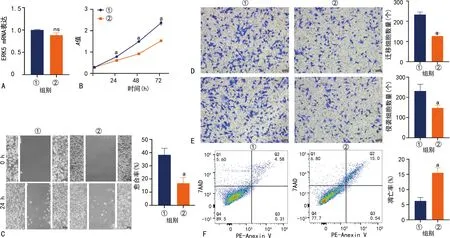
A:ERK5 mRNA表达水平;B:细胞增殖能力;C:划痕实验结果(100×);D:迁移实验结果(200×);E:侵袭实验结果(200×);F:流式细胞术分析结果;①:Control组;②:XMD17-109组;ns:P>0.05,与Control组比较;a:P<0.05,与Control组比较。
2.2 敲低与过表达ERK5对U251细胞的影响
与siNC组比较,siERK5组ERK5和ITPRIP mRNA和蛋白表达水平明显下调;与Vector组比较,ERK5-OE组ERK5和ITPRIP mRNA和蛋白表达水平明显上调;与siNC组比较,siERK5组U251细胞增殖、侵袭和迁移能力均明显降低,细胞凋亡率明显升高(P<0.05);与Vector组比较,ERK5-OE组U251细胞增殖、侵袭和迁移能力均明显升高(P<0.05),见图2~4。

A:8组ITPRIP mRNA表达水平;B:8组ERK5和ITPRIP蛋白Western blot结果;C:8组ERK5蛋白表达水平;D:8组ITPRIP蛋白表达水平;①Control组;②XMD17-109组;③siNC组;④siERK5组;⑤Vector组;⑥ERK5-OE组;⑦Vector+XMD17-109组⑧ERK5-OE+XMD17-109组;a:P<0.05,与Control组比较;b:P<0.05,与siNC组比较;c:P<0.05,与Vector组比较;d:P<0.05,与Vector+XMD17-109组比较。
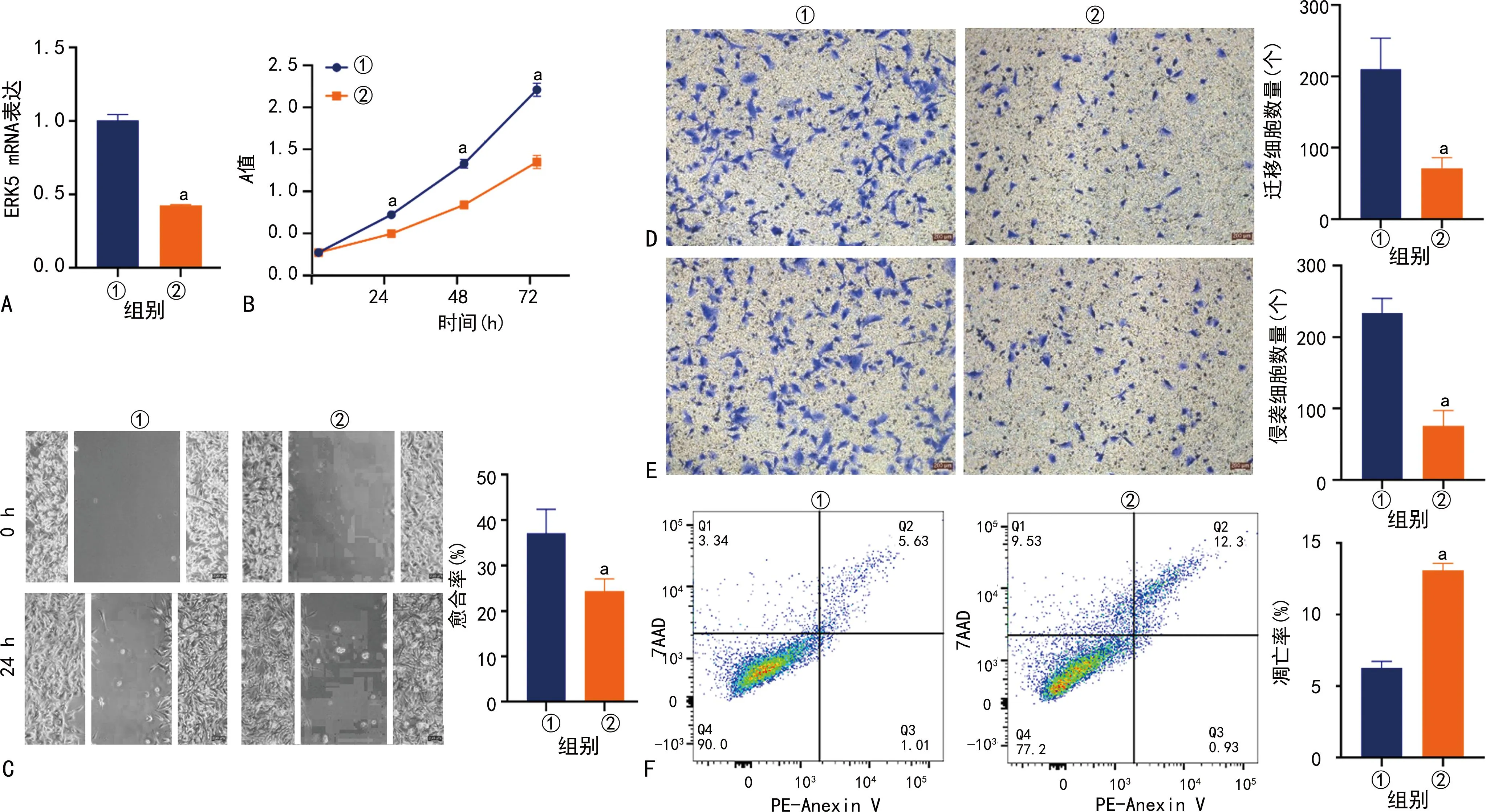
A:ERK5 mRNA表达水平;B:细胞增殖能力;C:划痕实验结果(100×);D:迁移实验结果(200×);E:侵袭实验结果(200×);F:流式细胞术分析结果;①:SiNC组;②:SiERK5组;a:P<0.05,与SiNC组比较。
2.3 XMD17-109逆转ERK5对U251细胞的调控作用
与Vector+XMD17-109组比较,ERK5-OE+XMD17-109组ERK5和ITPRIP mRNA和蛋白表达水平明显上调;与Vector+XMD17-109组比较,ERK5-OE+XMD17-109组U251细胞增殖、侵袭和迁移能力均明显升高,细胞凋亡率明显降低,见图2、5。
3 讨 论
原发或复发的胶质瘤起源于分化细胞,如成熟的神经元、星形胶质细胞和成体神经干细胞[20-21]。这一现象表明,恶性胶质瘤可能保留神经生物学特征和信号,使得神经中心胶质瘤治疗成为一种有前途的方法[22-24]。ERK5在调节多种凋亡信号通路中发挥重要作用,不仅促进肿瘤生长,还参与肿瘤细胞的增殖[25-26]。另一方面,ITPRIP缺陷已被证明会加重短暂性脑缺血和急性兴奋性毒性后的神经毒性[27]。基于这一认识,作者认为在胶质瘤细胞中,ERK5可能通过蛋白-蛋白相互作用对肿瘤启动子ITPRIP发挥促进作用,类似于其在神经元细胞中的作用。
ERK5在癌症中的致癌作用归因于其靶向特定的mRNA[28]。采用siRNA/shRNA沉默或敲除ERK5基因的多项研究表明,ERK5信号通路对多种细胞表型发挥重要调控作用。研究数据显示,体内和体外实验中沉默ERK5均能抑制多种癌细胞细胞的增殖作用,如子宫内膜癌、非小细胞肺癌、小细胞肺癌、黑色素瘤、三阴性乳腺癌和胆管癌等[29]。在小细胞肺癌中,沉默ERK5后细胞相较于野生型细胞增殖活力明显降低,这一结果强调了ERK5激酶活性在小细胞肺癌细胞增殖中的重要作用。此外,一项针对实验用增殖表皮癌细胞的研究显示,表皮生长因子能够引起ERK5的活化[30]。ERK5通过调节基质相关基因、促血管生成因子和整合素维持三阴性乳腺癌细胞的迁移和间质表型[17,31]。一项关于骨肉瘤的研究显示,ERK5是一种有效的预后指标,调节ERK5-STAT3通路会加剧骨肉瘤细胞的恶性表型[32]。
通过数据库对肿瘤基因表达谱进行分析,结果表明ERK5与ITPRIP在胶质瘤组织中的表达上调。基于此,作者推测ITPRIP在胶质瘤中的作用可能是通过调控ERK5轴介导。为证实这一假设,本研究进行了多项功能实验,发现XMD17-109处理后细胞表型明显抑制,ERK5表达无明显变化,ITPRIP表达降低。这是因为XMD17-109作为ERK5选择性抑制剂,通过阻断细胞内表皮生长因子诱导的ERK5自磷酸化,对ERK5下游分子发挥抑制作用,但其对ERK5本身无明显抑制作用。敲低ERK5会抑制胶质瘤细胞增殖、迁移和侵袭能力,诱导细胞凋亡,下调ITPRIP表达。相反,过表达ERK5对胶质瘤细胞的生物学功能起到促进作用,同时上调ITPRIP表达。ERK5抑制剂XMD17-109减弱了胶质瘤细胞增殖、迁移和侵袭能力,同时能够逆转过表达ERK5对胶质瘤细胞表型的促进作用。此外,前期研究证实,敲低ITPRIP对于胶质瘤细胞表型具有抑制作用[19],因此可以推论:XMD17-109通过阻断ERK5自磷酸化抑制ITPRIP表达,进而抑制胶质瘤细胞表型。
综上所述,本研究证实了XMD17-109通过抑制ERK5/ITPRIP信号通路进而抑制胶质瘤的进展,ERK5、ITPRIP可以作为胶质瘤预防、诊断和治疗的潜在生物标志物。
