丝状自组装蛋白支架介导的双酶级联催化体系构建促进D-塔格糖合成
2023-12-15李英刘伟朱丽英周治江凌
李英,刘伟,朱丽英,周治,江凌,4
(1.南京工业大学食品与轻工学院,江苏南京 210009)(2.南京工业大学化学与分子工程学院,江苏南京 210009)(3.南京工业大学生物与制药工程学院,江苏南京 210009)(4.南京工业大学材料化学工程国家重点实验室,江苏南京 210009)
D-塔格糖的甜度高达蔗糖的92%,但能量仅含蔗糖三分之一,具有降血糖[1,2]、改善肠道菌群[3]、抗氧化[4]的作用,在食品和医药领域有广泛应用。D-塔格糖的合成主要通过化学法和生物法。化学法[5]合成过程不易控制且副产物难以分离限制了其应用。目前,商业化的D-塔格糖通常是由D-半乳糖经过L-阿拉伯糖异构酶(L-AI,EC 5.3.1.4)的异构化生产[6]。然而,L-AI的固有热力学平衡以及D-半乳糖和D-塔格糖之间的性质相似,仍然存在转化率有限和纯化成本高的问题[7]。除了通过L-AI转化D-半乳糖途径外,还可通过半乳糖醇脱氢酶将半乳糖醇一步转化为D-塔格糖。Muniruzzaman等[8]使用Enterobacter agglomerans221e菌株生产D-塔格糖,半乳糖醇转化率高达92%。除了发酵法,Zhang等[9]通过在胞外偶联半乳糖醇脱氢酶和再生NAD+的NADH氧化酶,以100 mmol/L D-半乳糖醇为底物,12 h后的产率达90%。虽然利用D-半乳糖醇产D-塔格糖产率高,但是半乳糖醇价格昂贵,直接利用其做底物不符合经济效益。为降低底物成本,乳糖或富含乳糖的工业废物是研究者常用的廉价底物。Jia等[10]利用重组大肠杆菌粗酶液可实现以100 g/L乳糖的乳清粉为原料,制得23.50 g/L D-塔格糖。氧化还原反应转化率高,且没有复杂的副产物,是理想的途径。
有研究表明氧化还原反应中,辅因子限制是影响产量的重要因素[11]。半乳糖醇脱氢酶在将半乳糖醇转化为D-塔格糖的同时需要消耗辅因子。为避免大量添加外源辅因子且提高产量,多酶级联方式的利用具有重要意义。多酶级联催化已经成为生产高价值产物的重要方式[12-15],由于其“绿色”、高效、可持续等。同时,由于底物通道[16]或空间临近效应[17,18],可以使得一个酶的产物快速成为另一个酶的底物,加快反应速度[19]。基于蛋白质的生物分子支架是目前应用最广泛的多酶固定化材料之一,由于其可进行基因编辑,可由细菌合成、组装条件温和且经济[20-22]。Clark等[23]将从深海超嗜热菌詹氏甲烷球菌(Methanocaldococcus jannaschii)中分离出的预折叠蛋白的单个亚基γ-PFD(γ-Prefoldin)突变成可通过静电相互作用有序排列的EE、KK单体,单体的N-或C-端很容易设计来连接功能分子。并利用了荧光蛋白对(mCerulean3、mVenus),证明了其可在胞外自组装[24],表明其有作为支架固定酶的潜力。然而,并未进一步探究其胞内应用。
基于此,在大肠杆菌中异源表达树干毕赤酵母(Scheffersomyces stipitisCBS 6054)的D-木糖还原酶(SsXR)和豌豆根瘤菌(Rhizobium leguminosarumbv.viciae3841)来源的半乳糖醇脱氢酶(RlGDH),通过大肠杆菌内源性β-半乳糖苷酶将乳糖转化为半乳糖,SsXR将半乳糖转化为半乳糖醇,RlGDH将其进一步转化为D-塔格糖。为降低中间产物的流失,利用蛋白支架EE、KK分别与和SsXR、RlGDH进行融合,依靠支架的静电相互作用实现双酶级联,提高D-塔格糖合成效率(图1),并对其摇瓶中发酵条件进行优化探究,以期为D-塔格糖的高效生物合成提供潜在菌株。
1 材料与方法
1.1 试剂和材料
1.1.1 菌株和质粒
DH5α、BL21大肠杆菌感受态购买自生工生物工程(上海)有限公司,本研究用菌株和质粒见表1。
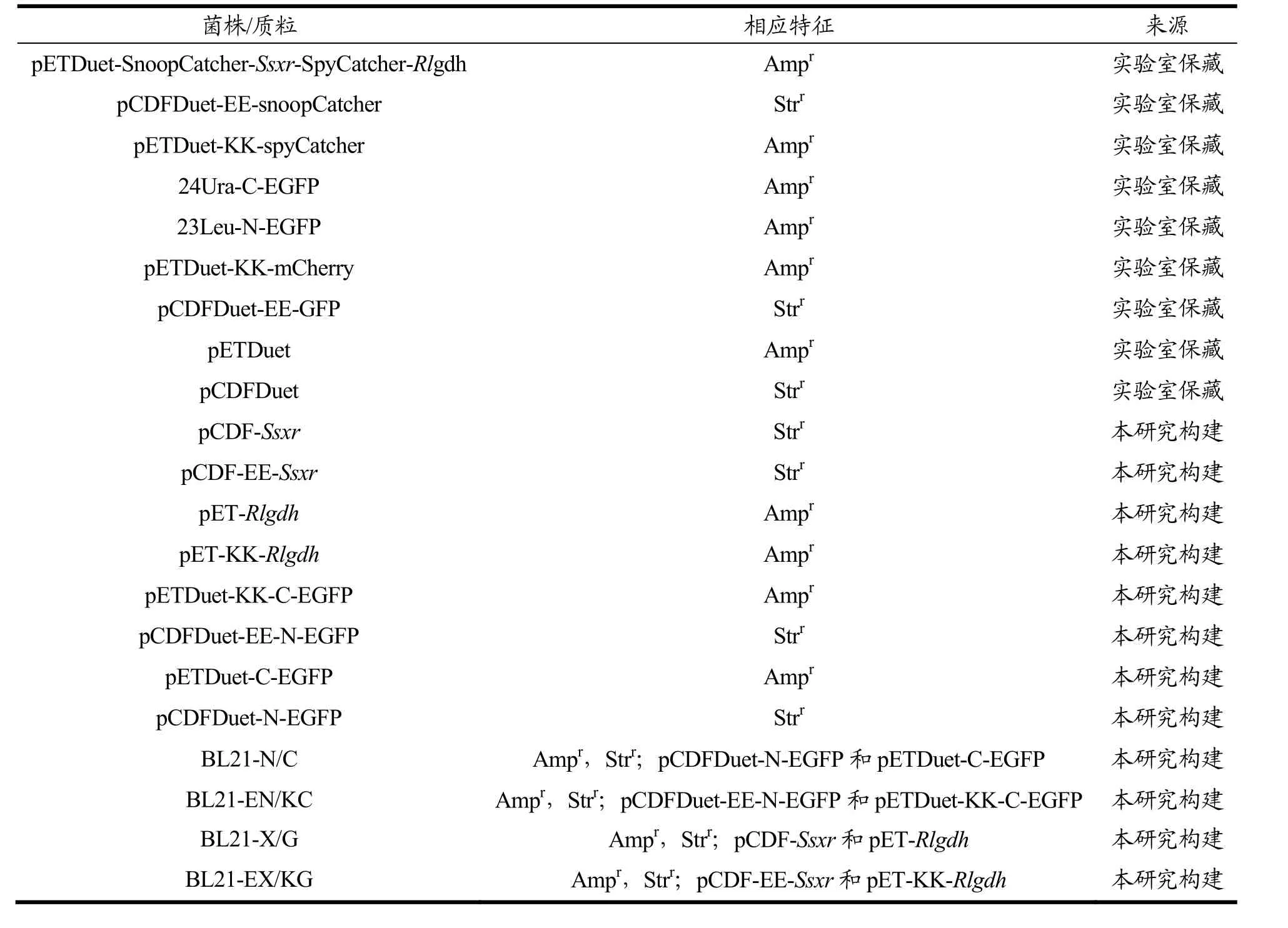
表1 本研究所用的质粒和菌株Table 1 Strains and plasmids used in this study
1.1.2 主要生化试剂
蛋白胨、酵母粉,OXOID;琼脂粉、氯化钠,国药集团;琼脂糖、D-塔格糖、L-半胱氨酸盐酸盐无水物,源叶生物科技有限公司;乳糖、咔唑,上海麦克林生化科技股份有限公司;硫酸,永华化学科技(江苏)有限公司;磷酸二氢钠、磷酸氢二钠,西陇科学股份有限公司;BamHI、XhoI等限制性核酸内切酶购于TaKaRa公司;Phanta Max Super-Fidelity DNA Polymerase、小量质粒提取试剂盒、同源重组试剂盒和胶回收试剂盒购于南京诺维赞生物科技有限公司;Ni预装重力柱、变性丙烯酰胺凝胶快速制备试剂盒,生工生物工程(上海)股份有限公司。
Buffer A:20 mmol/L咪唑、500 mmol/L氯化钠、20 mmol/L磷酸钠缓冲液,pH值8.00。
Buffer B:300 mmol/L咪唑、500 mmol/L氯化钠、20 mmol/L磷酸钠缓冲液,pH值8.00。
LB液体培养基(g/L):蛋白胨10、酵母粉5、氯化钠10。
LB固体培养基(g/L):蛋白胨10、酵母粉5、氯化钠10、琼脂粉20。
TB培养基(g/L):蛋白胨12、酵母粉24、甘油10、磷酸二氢钾2.30、磷酸氢二钾12.50。
M9培养基(g/L):七水合磷酸二氢钠12.80、磷酸二氢钾3、氯化钠0.50、氯化铵1、1 mol/L MgSO42 mL、20%葡萄糖溶液20 mL、1 mol/L氯化钙0.10 mL,补充水至1 L。
1.2 主要仪器与设备
MQT-60R摇床,上海旻泉仪器有限公司;SHP-150生化培养箱,上海精宏实验设备有限公司;HH-S2数显恒温水浴锅,江苏金怡仪器科技有限公司;GL-20G-II台式冷冻离心机,上海安亭科学仪器厂;HN98超声波信号发生器,上海汗诺仪器有限公司;T960A智能PCR仪,杭州晶格科学仪器有限公司;721N紫外分光光度计,上海菁华科技仪器有限公司;核酸电泳仪,上海天能科技有限公司;F-7000荧光光谱仪,日立公司;科乐比微量分光光度计,德国伯赫。
1.3 方法
1.3.1 重组大肠杆菌的构建
以本实验室保藏的质粒为模版,参照NCBI获得的SsXR、RlGDH(GenBank AcceSsion No.分别为XP_001385181、WP_011650422)和EE/KK基因序列设计引物进行PCR扩增,如表1所示,引物BamH I-SsXR-F和SsXR-XhoI-R、BamH I-RlGDH-F和RlGDH-XhoI-R所对应的模板为质粒pETDuet-SnoopCatcher-SsXR-SpyCatcher-RlGDH分别扩增片段SsXR、RlGDH;引物r-SsXR-F和SsXR-XhoI-R(r-RlGDH-F和RlGDH-XhoI-R)以pETDuet-SnoopCatcher-SsXR-SpyCatcher-RlGDH质粒为模版,pET-BamH I-KK-F和KK-R(pCDF-BamH I-EE-F和EE-R)所对应的模板为质粒pETDuet-KK-spyCatcher(pCDFDuet-EE-snoopCatcher);获取的片段通过引物pET-BamH I-KK-F(pCDF-BamH I-EE-F)和SsXR-XhoI-R(RlGDH-XhoI-R)融合pcr扩增得到KK-SsXR(EE-RlGDH)。PCR反应条件如下:95 ℃预变性5 min,95 ℃变性30 s,退火30 s,退火温度根据引物Tm值设定,72 ℃延伸,延伸时间根据序列长度设定,33个循环后在72 ℃延伸5 min,4 ℃保存。PCR产物通过0.80%的琼脂糖凝胶电泳验证。SsXR、RlGDH、KK-SsXR和EE-RlGDH片段通过BamH I和XhoI双酶切连接到载体上(pETDuet、pCDFDuet)。通过T4连接酶16 ℃过夜连接并导入DH5α大肠杆菌感受态细胞中。利用抗性基因平板筛选进行菌落PCR验证,提取质粒送至通用生物(安徽)股份有限公司进行测序,利用snapgene软件进行同源比对,确定插入序列正确性,得到正确的质粒pCDF-SsXR、pCDF-EE-SsXR、pET-RlGDH、pET-KK-RlGDH。最后将提取得到的正确质粒转化大肠杆菌BL21感受态细胞得到重组菌(对照组:BL21-X/G、实验组:BL21-EX/KG)。
用相同的方法,利用引物pET-KK-gj-R和pET-KK-gj-F,pCDF-EE-gj-R和pCDF-EE-gj-F分别以pET-KK-RlGDH和pCDF-EE-SsXR为模版得到片段pET-KK-gj和pCDF-EE-gj,引物C-EGFP-F和C-EGFP-R,N-EGFP-F和N-EGFP-R分别以24Ura-C-EGFP和23Leu-N-EGFP为模版获得片段C-EGFP和N-EGFP。利用同源重组连接并导入DH5α大肠杆菌感受态细胞中。获得质粒pETDuet-KK-C-EGFP、pCDFDuet-EE-N-EGFP。引物BamHI-C-EGFP-F和XhoI-C-EGFP-R,BamHI-N-EGFP-F和XhoI-N-EGFP-R分别以24Ura-C-EGFP和23Leu-N-EGFP为模版获得片段C-EGFP和N-EGFP通过双酶切连接到载体上,利用通过T4连接酶16 ℃过夜连接并导入DH5α大肠杆菌感受态细胞中,通过验证得到pETDuet-C-EGFP和pCDFDuet-N-EGFP。最后将提取得到的正确质粒转化大肠杆菌BL21感受态细胞得到重组菌游离组(BL21-N/C)和支架组(BL21-EN/KC)。本研究使用的引物序列参见表2。
1.3.2 蛋白支架胞内外表征
1.3.2.1 EE-mCherry和KK-GFP酶胞外自组装表征
有研究表明能观察荧光共振现象只能是两种荧光蛋白距离小于10 nm时[25]。因此,利用已经构建的大肠杆菌BL21/pCDF-EE-mCherry、BL21/pET-KK-GFP,将其在37 ℃培养OD600到0.60~0.80,加入0.25 mmol/L IPTG,20 ℃诱导24 h后,6000 r/min 10 min收集菌体,用50 mmol/L Tris HCl(pH值7.0)清洗三次,重悬超声破碎,12000 r/min 4 ℃离心10 min上清液通过Ni柱纯化,通过Buffer A去除杂蛋白后,Buffer B洗脱目标蛋白,移入透析袋置于纯水中透析去除高浓度盐离子。利用SDS PAGE验证重组蛋白的可溶性表达,纯化的蛋白浓度通过科乐比微量分光光度计测定。将透析得到的纯蛋白EE-mCherry和KK-GFP以1:0、1:0.5、1:1、1:2、1:4、1:12在50 mmol/L PBS(pH值7.0)混合室温下自组装1 h后,荧光光谱仪测定荧光强度。
1.3.2.2 KK-C-EGFP和EE-N-EGFP荧光蛋白胞内自组装表征
分裂成两部分的C-EGFP和N-EGFP荧光蛋白,只有当两个分裂组分被靶向靠近时,才能观察到荧光强度的增强,广泛应用在表征胞内的自聚集效果[26,27]。因此,将重组菌(游离组:BL21-N/C和支架组:BL21-EN/KC)接种于5 mL LB,添加5 μL氨苄青霉素(100 mg/mL)和5 μL硫酸链霉素(50 mg/mL),37 ℃过夜培养。以2%(V/V)的接种量接种于50 mL TB培养基中,37 ℃、200 r/min培养至OD600约为0.60~0.80时,添加终浓度为0.25 mmol/L的IPTG于20 ℃下诱导24 h,取适量菌液离心、得到菌体用50 mmol/L Tris-HCl缓冲液(pH值7.0)洗涤菌体3次,重悬,通过稀释得到OD600相近的菌体重悬液,以488 nm为激发波长,518 nm为发射波长测定荧光强度。
1.3.3 重组大肠杆菌发酵合成D-塔格糖方法
从活化平板上挑取单菌落(游离组、支架组)接种至5 mL LB液体培养基中,添加相应的抗性,37 ℃、160 r/min振荡培养过夜。以2%(V/V)的接种量,接种于含10 g/L乳糖的50 mL LB培养基中,37 ℃、200 r/min培养至OD600约为0.60~0.80时,添加终浓度为0.25 mmol/L的IPTG于25 ℃下诱导与发酵培养。在规定的时间点取样,并测定D-塔格糖产量。
1.3.4 摇瓶发酵条件优化
1.3.4.1 发酵培养基种类优化
选择TB、M9以及LB培养基,其余条件不变,按照1.3.3进行发酵。
1.3.4.2 发酵温度优化
以LB为培养基,设置诱导与发酵温度为16、20、25、30、37 ℃,其余条件不变,按照1.3.3进行发酵。
1.3.4.3 诱导剂添加量优化
以LB为培养基,诱导与发酵温度为20 ℃,设置诱导剂添加量为0.1、0.25、0.25、1 mmol/L,其余条件不变,按照1.3.3进行发酵。
1.3.5 D-塔格糖与乳糖测定方法
发酵液上清经0.22 μm滤膜过滤后,采用Carbomix Ca-NP10(10 μm,交联度8%,7.80×300 mm)柱的岛津高效液相色谱仪(示差折光检测器)分析测定D-塔格糖与乳糖。流动相为100%水,流速0.40 mL/min,85 ℃,洗脱35 min。
1.3.6 数据分析
结果以实验的均数±标准差(mean±SD)表示,每次实验3次重复。采用Microsoft Excel 365软件计算平均值和SD值;采用SPSS26.0进行单因素方差分析确定显著性差异,n.s无显著性差异(P>0.05)、*差异显著(P<0.05)、**差异非常显著(P<0.01)****差异极显著(P<0.0001),并用Origin 2018进行绘图。
2 结果分析
2.1 EE/KK蛋白支架表征
2.1.1 EE-mCherry和KK-GFP酶胞外自组装表征
为探究EE/KK支架是否具有自组装特性,首先在胞外进行表征。将荧光蛋白KK-mCherry、EE-GFP通过镍柱纯化,SDS PAGE结果表明KK-mCherry、EE-GFP可以形成良好的可溶性表达(图2)。以不同的化学计量比混合在50 mmol/L PBS(pH值7.0)孵育1 h后,测定得到的荧光强度为y轴和波长为x轴得到图3。图3表明,随着KK-mCherry的量增加,GFP在510 nm的荧光强度逐渐降低降低,而KK-mCherry在610 nm处的荧光强度逐渐增加。这表明荧光蛋白对形成了荧光共振,证实EE/KK支架可以胞外自组装。
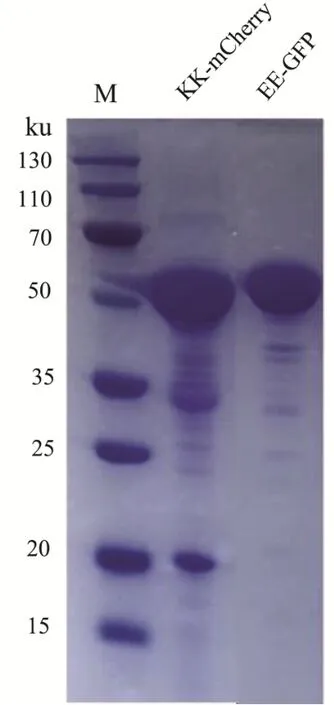
图2 纯化的重组蛋白KK-mCherry和EE-GFP的SDS PAGEFig.2 SDS PAGE of purified recombinant proteins KK-mCherry and EE-GFP

图3 以不同摩尔比组装的EE-GFP和KK-mCherry荧光蛋白复合物的荧光发射光谱Fig.3 Fluorescence emission spectra of EE-GFP and KK-mCherry fluorescent protein complexes assembled at different molar ratios
2.1.2 KK-C-EGFP和EE-N-EGFP荧光蛋白胞内自组装表征
前面成功验证了支架可在胞外自组装。为进一步验证支架在胞内的自组装特性,将构建的重组大肠杆菌游离组(BL21-N/C)和支架组(BL21-EN/KC)分别诱导,使得荧光蛋白(C-EGFP、N-EGFP)和支架-荧光蛋白复合体(KK-C-EGFP、EE-N-EGFP)在胞内表达。取诱导液离心收集菌体。稀释得到相同OD600的菌液,观察到支架组的荧光强度增加了约13.33倍(图4)。Lau等[26]在酿酒酵母中通过包封蛋白实现分裂的黄色荧光蛋白的荧光强度仅增大2.5倍。紫外灯照射下,可见支架组的荧光强度显著高于游离组(图5)。以上结果表明,EE/KK支架可在胞内成功自组装,实现目标蛋白的级联。

图4 游离组和支架组荧光强度对比图Fig.4 Comparison chart of fluorescence intensity between the free group and the scaffold group

图5 紫外光下的游离组(BL21-N/C)和支架组(BL21-EN/KC)菌液荧光对比图Fig.5 Fluorescence comparison of bacteria solution in the free group (BL21-N/C) and scaffold group (BL21-EN/KC) under ultraviolet light
2.2 EE、KK自组装支架用于D-塔格糖合成
基于EE、KK支架的胞内表征实验结果,进一步考察其在胞内组装氧化还原途径双酶合成D-塔格糖的能力。以大肠杆菌E. coliBL21为出发菌株,选取Scheffersomyces stipitisCBS 6054来源的木糖还原酶(SsXR)以及Rhizobium legumenosarum来源的半乳糖醇脱氢酶(RlGDH)催化D-半乳糖合成D-塔格糖。以构建的重组质粒pCDF-SsXR与pET-RlGDH,进而构建游离酶菌株BL21-X/G,SDS-PAGE结果显示SsXR(35.9 ku)与RlGDH(27.3 ku)成功表达(图6,泳道1)。分别将EE、KK使用GSlinker(GGGGSGGGGS)连接至SsXR与RlGDH的N端,构建重组质粒pCDF-EE-SsXR与pET-KK-RlGDH,进而构建支架级联型菌株BL21-EX/KG,SDS-PAGE结果显示EE-SsXR(56.0 ku)与KK-RlGDH(47.3 ku)成功表达(图6,泳道2)。

图6 E. coli BL21-X/G与E. coli BL21-EX/KG中蛋白表达SDS-PAGEFig.6 SDS-PAGE of protein expression in E. coli BL21-X/G and E. coli BL21-EX/KG
在含有10 g/L乳糖的LB培养基中,重组菌株BL21-X/G与BL21-EX/KG培养至OD600为0.50~0.60时,发酵温度由37 ℃调至25 ℃,加入0.25 mmol/L IPTG进行诱导。发酵48 h,EE-KK支架级联体系(BL21-EX/KG)的D-塔格糖产量达到3.52 g/L,相比于游离体系(BL21-X/G)提高50%(图7)。结果说明,基于EE/KK的蛋白支架可以有效级联胞内目标酶,强化催化效率。据此推测,EE-KK蛋白支架可能通过拉进SsXR与RlGDH酶之间的距离,产生类似“底物通道”效应[16],此外,SsXR与RlGDH催化所需的辅酶(NAD(P)H/NAD(P)+)之间也可以快速循环,进而提高D-塔格糖的合成效率。

图7 游离酶体系与EE-KK支架级联体系合成D-塔格糖产量比较Fig.7 Comparison of the titer of D-tagatose synthesis by free enzyme system and EE/KK scaffold cascade system
2.3 摇瓶发酵条件优化
进一步地,通过优化发酵培养基种类、发酵温度、IPTG添加量,以期BL21-EX/KG在摇瓶发酵合成D-塔格糖的产量得到进一步提升。
2.3.1 发酵培养基种类优化
结果如图8所示,发酵48 h,M9培养基中菌株生物量(OD600为8.12)与D-塔格糖产量最低(1.07 g/L),这可能因为M9培养基成分简单,有机氮源较为缺乏,影响菌体的生长以及蛋白的表达[28]。而利用TB培养基和LB培养基,48 h菌株生物量分别达到12.30和11.10。虽然TB培养基略高于LB培养基,但是TB培养基中D-塔格糖的产量却显著低于LB培养基(1.85 g/L VS 3.52 g/L)。猜测是因为TB培养基中含有甘油,由于甘油的摄入与代谢会提高大肠杆菌胞内NADH的含量,消耗NAD+[29],而RlGDH在催化半乳糖醇合成D-塔格糖的同时也会消耗NAD+合成NADH,因此,甘油的添加可能会导致NAD+的供给不足,进而影响D-塔格糖的合成。

图8 发酵培养基种类对D-塔格糖合成的影响Fig.8 Effect of medium type on D-tagatose synthesis
为了进一步验证我们的猜测,在LB培养基中分别添加10 g/L甘油、10 g/L葡萄糖以及微量元素,考察对BL21-EX/KG合成D-塔格糖的影响。结果如图9所示,微量元素的添加对D-塔格糖的合成具有一定的积极影响,产量略有提高。葡萄糖的添加使得D-塔格糖产量降低56%,这归结于大肠杆菌中存在的“葡萄糖效应”,影响乳糖的转运和代谢[30]。而甘油的添加也降低了37%的D-塔格糖产量,这与在利用TB培养基发酵合成过程中的现象一致,证实了我们的猜测,LB培养基中,甘油的添加会影响D-塔格糖的合成。因此,后续的摇瓶发酵培养基选用LB培养基。

图9 LB发酵培养基中添加甘油、葡萄糖以及微量元素对D-塔格糖合成的影响Fig.9 Effects of adding glycerol、glucose and trace elements in LB fermentation medium on the synthesis of D-tagatose
2.3.2 发酵温度与诱导剂添加量优化
温度和IPTG浓度是影响外源蛋白表达的重要因素,此外发酵温度也影响细胞生长代谢与产物的合成。我们在LB培养基中进一步考察了不同发酵温度和IPTG添加量对合成D-塔格糖的影响。
首先考察了温度对D-塔格糖合成的影响。图10所示,菌体生物量随着发酵温度的升高而增加,30 ℃与37 ℃时菌体生长速率较快,在16 ℃下则相反。不同的是,D-塔格糖的产量与温度呈负相关。在20 ℃下,D-塔格糖产量最高,达到3.82 g/L。可能是较高的温度影响蛋白的表达,易形成包涵体。因此,综合考虑,后续选择20 ℃为D-塔格糖合成的发酵温度。

图10 不同发酵温度对D-塔格糖合成的影响Fig.10 Effects of different fermentation temperatures on the synthesis of D-Tagatose
除温度之外,IPTG添加量对蛋白的表达也同样重要。我们进一步比较了不同IPTG浓度(0.1、0.25、0.5和1.0 mmol/L)对D-塔格糖合成的影响。结果如图11所示,随着IPTG浓度的升高反而导致D-塔格糖产量的降低。这与Couto等[31]研究结果一致,较高的IPTG浓度不会导致效价提高反而会造成代谢负荷从而影响生产力。0.1和0.25 mmol/L的添加量下,D-塔格糖的产量无显著性差异。因此,选择0.1 mmol/L作为后续实验的IPTG添加量。
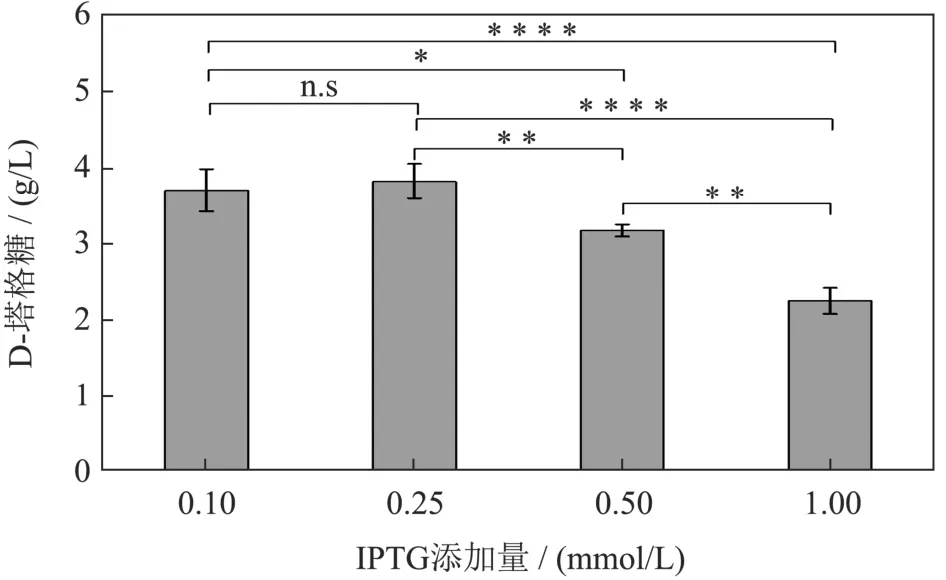
图11 不同IPTG浓度对D-塔格糖合成的影响Fig.11 Effects of different IPTG concentrations on the synthesis of D-Tagatose
2.3.3 最适摇瓶发酵条件下菌株BL21-EX/KG合成D-塔格糖
基于上述优化条件,在摇瓶中考察菌株BL21-EX/KG批次发酵合成D-塔格糖的能力,结果如图12所示。发酵48 h后,菌体到达稳定期,OD600为11.50。D-塔格糖的产量持续增长,到60 h时到达峰值3.93 g/L,此时乳糖几乎完全消耗,D-塔格糖对乳糖的转化率达到0.39 g/g,为理论转化率(0.53 g/g)的74%。Zhang等[32]通过肽接头(GGGGS)3偶联β-D-半乳糖苷酶和L-阿拉伯糖异构酶实现从乳糖产D-塔格糖,转化率为42.40%。Zhang等[33]在半乳糖激酶基因失活的植物乳杆菌中过表达β-半乳糖苷酶和L-阿拉伯糖异构酶,实现33%的乳糖转化率。可知,本实验构建的菌株可实现较高的乳糖转化率。然而乳糖消耗速率较慢,从而导致较低的D-塔格糖生产强度[0.07 g/(L·h)]。猜测可能与摇瓶中较低的生物量有关。此外,大肠杆菌通过乳糖透性酶转运乳糖,转运效率较低。因此,后续实验可以结合高密度发酵以及细胞透性化[34]提高D-塔格糖的生产强度。
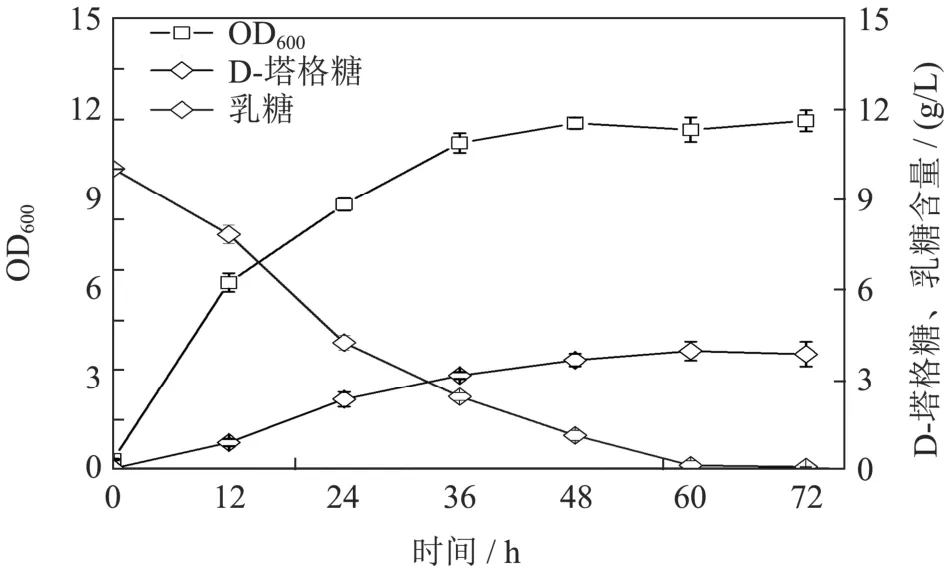
图12 BL21-EX/KG摇瓶发酵合成D-塔格糖过程Fig.12 D-Tagatose production from fermentation by BL21-EX/KG
3 结论
本研究首先验证了EE-KK单体在胞外和胞内均具有自组装能力。为进行概念验证,以大肠杆菌BL21为底盘细胞,通过与自组装蛋白支架EE、KK融合表达SsXR、RlGDH,成功实现多酶级联催化乳糖合成D-塔格糖。支架组BL21-EX/KG相较于游离组BL21-X/G,塔格糖产量提高了50%。进一步从发酵培养基种类、IPTG添加量、发酵温度优化了发酵条件,得到最佳发酵条件为:LB培养基,温度20 ℃,0.1 mmol/L IPTG添加量。以10 g/L乳糖为底物,摇瓶中BL21-EX/KG可实现D-塔格糖产量达到3.93 g/L,为理论转化率的74%。本研究为基于氧化还原酶途径生物合成D-塔格糖提供了研究基础,为多酶级联组装提供了有效的工具。
