遗传性出血性毛细血管扩张症合并肺动脉高压1例
2023-12-14樊铭薇程哲堕梦洁刘青赵文静
樊铭薇 程哲 堕梦洁 刘青 赵文静
遗传性出血性毛细血管扩张症(HHT),也称为Rendu-Osler-Weber综合征,是一种罕见的常染色体显性遗传血管疾病,其合并肺动脉高压的病例更为少见,所以对于该病的治疗方案和预后仍需大量研究进行探索,为该病提供更多的理论依据来指导临床工作,特报道此病例为其提供临床诊疗方案。
病例资料
患者,女,31岁,2022年3月5日因“劳力性胸闷、喘憋感2年,加重15天”就诊于我院,2年前出现劳力性胸闷、喘憋感,表现为劳累后加重,休息后缓解,并伴双下肢及颜面部间断性水肿,晨轻暮重,无头晕、头痛、咳嗽、咳痰、咯血不适,曾就诊于三门峡市中心医院,诊断“肺动脉高压”,予“西地那非”靶向治疗,未规律服药,症状反复。15天前上述症状加重,伴不规则发热,最高可达38.5 ℃,就诊于当地医院,pro-BNP:7291.47 pg/mL,超声心动图示:重度肺动脉高压、肺动脉内径增宽、右房、右室增大、三尖瓣大量返流、肺动脉瓣少量返流,予“西地那非、安利生坦”降肺动脉压,疗效不佳。发病以来,食欲欠佳。追问病史得知患者自13岁起有反复鼻出血史,其父亲及1兄也有鼻出血病史。入院查体:轻度贫血貌,舌体表面可见散在出血点,其父亲舌体也可见出血点(图1),呼吸急促,肋间隙正常,语颤正常,未闻及干湿啰音,腹部膨隆,双下肢及颜面部可见中度水肿。
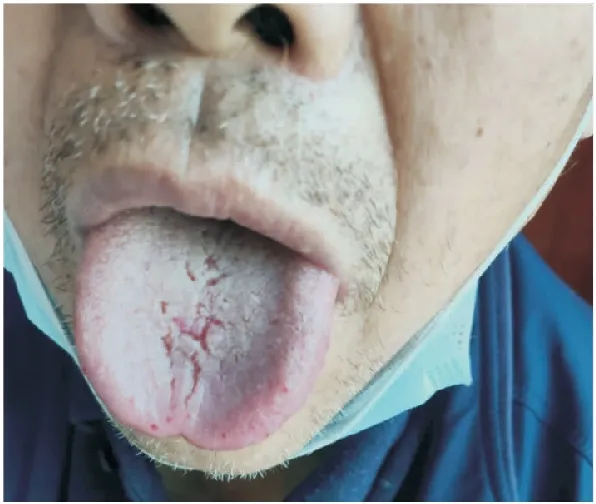
图1 患者父亲舌体可见出血点
入院完善检查,血常规提示轻度贫血;尿常规:隐血3+,蛋白2+;粪便常规:隐血阳性;心肌酶:LDH 280 U/L、cTnT 0.024 ng/mL、TnI 0.023 μg/L、pro-BNP 4969pg/mL;D-二聚体1.24 mg/L;ESR 24 mm/h、CRP 9.86 mg/L、PCT 0.242 ng/mL;肾功能:尿素氮19.2 mmol/L、肌酐256μmol/L、尿酸560μmol/L、胱抑素C 2.3 mg/L;肝功能:谷氨酰转肽酶111 U/L、碱性磷酸酶147 U/L、5′核苷酸酶12.7 U/L、前白蛋白115 mg/L、总胆红素40.6 μmol/L、直接胆红素23.6 μmol/L、间接胆红素17 μmol/L;血脂:HDL 0.7 mmol/L;风湿免疫:抗J0-1抗体弱阳性;尿总蛋白浓度0.58 g/L、尿白蛋白浓度273.6 mg/L;甲功(-)、糖化血红蛋白(-);ECG:1.窦性心律不齐;2.完全性右束支传导阻滞;3.ST-T段异常改变(ST段Ⅱ、Ⅲ、aVF、V3-V6段压低;T波Ⅱ、Ⅲ、aVF、V1-V6导联倒置)。腹部彩超示:肝内异常血流(考虑广泛性动静脉瘘)、脾大、脾静脉增宽。心脏彩超示:左房径34 mm、左室径43 mm、右室径50 mm、肺动脉环径32 mm、EF 63%、肺动脉收缩压64 mmHg,超声诊断:重度肺动脉高压。肺动脉CTA(见图2~5)可见肺动脉主干增粗、右房及右室增大、右中肺及纵隔见迂曲血管汇入奇静脉、三支肝静脉增粗、下腔静脉增粗,继而完善右心声学造影提示肺动静脉瘘。6 min步行实验:299m。并于2022年3月21日行右心导管检查,肺动脉压力70/28 mmHg (40 mmHg),取动脉血测血气分析,肺动脉血氧饱和度69.1%,心排量6.9 L/min,术中予万他维20ug雾化吸入后,再测相关部位压力:右心房压27/9 mmHg(15 mmHg)、右心室压力71/4 mmHg(26 mmHg),肺动脉压力70/28 mmHg (40 mmHg),并取动脉血测血气分析,上腔静脉氧饱和度49.8%,右心房氧饱和70%,右心室氧饱和71.9%,心排量11.9 L/min,结合患者既往反复鼻出血病史、舌部毛细血管扩张、肺动静脉瘘,临床诊断为:遗传性岀血性毛细血管扩张症;右心漂浮导管确诊肺动脉高压,予吸氧、呋塞米、螺内酯利尿、西地那非0.25片tid、安立生坦10 mg qd降低肺动脉高压、纠正心衰、治疗贫血、升血小板、升白细胞等药物应用后,治疗后患者呼吸困难症状较前明显减轻,2022年3月22日出院,出院后嘱患者规律应用利尿剂、铁剂、肺动脉高压靶向药物治疗,规律郑大一附院呼吸内科随诊。
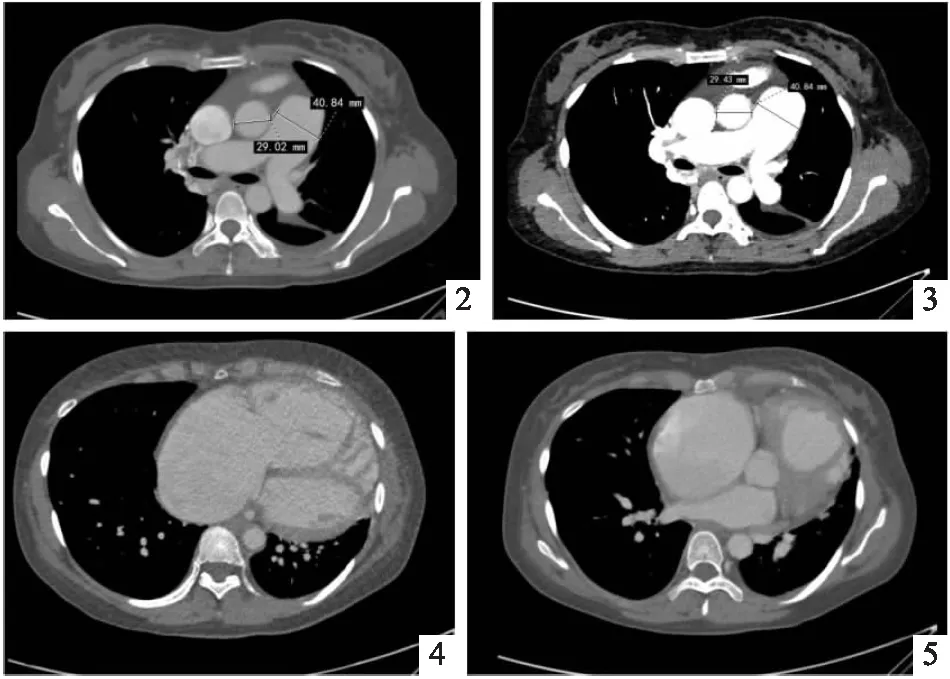
图2~5 (2022年3月6日)肺动脉CTA示肺动脉主干增粗,较宽处径约41 mm,右房、右室增大
讨 论
遗传性出血性毛细血管扩张症[1],发病率约1/5000,特征是皮肤、粘膜和或内脏器官出现多发性动静脉畸形。临床诊断基于四项库拉索标准:(1)反复自发鼻出血。(2)毛细血管扩张位于多个特征性部位:嘴唇、口腔、手指、鼻腔。(3)胃肠道的毛细血管扩张,肺、肝、脑或脊髓的动静脉畸形。(4)一级亲属根据以上标准被诊断为HHT,确诊标准:符合3条及3条以上;疑诊标准:符合2条;排除标准:0或1条标准,本例患者既往反复出现自发性鼻岀血,舌体表面毛细血管扩张,肺及肝脏存在动静脉瘘,符合确诊标准,明确诊断为HHT。反复自发性鼻出血是HHT的最常见症状,常会导致缺铁性贫血,约50%的患者在20岁之前出现鼻出血,肺动静脉畸形在HHT患者中发病率约 15%~50%,是威胁生命的严重并发症,本文报道病例存在贫血、鼻出血及肺动静脉瘘。
有研究[2]表示HHT的发病与基因突变相关,目前所有已知的导致HHT的基因缺陷都是在TGF-β信号通路中发现的,发现的突变基因有激活素A受体Ⅰ型(ACVRL1或ALK1)、内皮胶(ENG)、生长分化因子2(GDF2,也称为BMP9或SMAD9)或SMAD家族成员4(SMAD4)。有病例报道[3]HHT-PAH患者行基因检测出突变基因ACVRL1,此基因是目前发现HHT的常见突变基因。HHT中某些突变基因可能与特定临床表现相关,ENG基因突变可能与肺及脑动静脉畸形相关,ALK-1基因突变可能导致肝脏动静脉畸形、脊髓动静脉畸形、鼻出血、肺动脉高压及青少年息肉病[4],有相关病例[5]报道也证实了ACVRL1基因外显子9存在c.1313T>C(p.M438T)杂合错义突变,此病例中的患者因为家庭条件的限制未能行基因水平检测,但是追问病史得知患者自13岁起有反复鼻出血病史,并且其父亲及1兄也有鼻出血病史,彩超提示肝内广泛性动静脉瘘,因此不能排除患者基因检测异常的可能。
肺动脉高压(pulmonary hypertension,PH)是指在海平面、静息状态下,经右心导管检查测定的肺动脉平均压≥25 mmHg。《中国肺动脉高压诊断与治疗指南(2021版)》[6]指出部分PH发病也与基因突变相关,且遗传性 PAH(heritable pulmonary arterial hypertension,HPAH)均为单基因常染色体显性遗传,所以指南建议筛查与PAH相关的基因及高危人群,基因检测有助于 PAH家系成员明确自身是否携带致病突变基因,HHT的发病也与基因突变相关,所以携带突变基因但尚无临床表现的家族成员应早期筛查及密切随访,以便起到早期预防的作用。
据报道[7]肺动脉高压(PAH)是HHT的一种罕见表现,其在HHT人群中的确切患病率尚未阐明,PAH也是HHT的严重并发症,与HHT相关的PAH的临床特征及预后研究也十分有限,所以HHT-PAH的病例在临床上是相对罕见的。由于目前没有其长期研究,HHT-PAH患者的临床管理经验又知之甚少,且该病预后不良,所以对于该病的治疗方案和预后仍需大量研究进行探索,为该病提供更多的理论依据来指导临床工作。有队列研究[8]表示HHT-PAH患者的一年和三年生存率分别为77.8%和53.3%,并且总胆红素(TBil)可能是HHT-PAH患者长期预后的预测指标,TBil较高的HHT-PAH患者预后较差。本例HHT-PAH患者总胆红素40.60μmol/L,因此继续追踪患者1年及3年预后具有重要意义。
综上所述,遗传性出血性毛细血管扩张症合并肺动脉高压是相对罕见的疾病,发病率低,特报道此病例为HHT-PAH提供临床诊疗方案。
