AGPAT2基因突变致先天性全身性脂肪营养不良伴发疹性黄瘤1例
2023-12-11罗云云张沥元王心怡杜函泽
罗云云,张沥元,王心怡,刘 赫,杜函泽,潘 慧
中国医学科学院 北京协和医学院 北京协和医院 内分泌科,北京100730
先天性全身性脂肪营养不良(congenital generalized lipodystrophy, CGL)是一种常染色体隐性遗传病,临床表现为全身脂肪组织缺失、肌肉组织明显,可出现多毛、黑棘皮,继发糖代谢和脂代谢障碍[1]。目前已确定了至少4种分子结构不同的先天性脂肪营养不良类型,分别是CGL1型、CGL2型、CGL3型和 CGL4型。CGL1型为1酰基甘油3 磷酸O酰基转移酶2(1-acylglycerol-3-phosphate O-acyltransferase 2,AGPAT2)基因突变所致,CGL2型为Bernardinelli-Seip先天性脂肪代谢障碍2(Bernardinelli-Seip congenital lipodystrophy 2,BSCL2)基因突变所致,CGL3型为Ⅰ型窖蛋白(caveolin-1,CAV1)基因纯合无义突变所致,CGL4型由聚合酶Ⅰ和转录本释放因子(polymerase-1 and transcript release factor,PTRF)基因突变引起。其中CGL1和CGL2型最常见,占比CGL的95%[2],CGL2表型最严重,可伴有智力障碍、心肌损害及轻至中度精神问题[3],CGL1、3、4型机械性脂肪组织不受累,CGL3和CGL4型骨髓脂肪存在,CGL3型表现为身材矮小,CGL4型患者表现为全身性肌萎缩和轻度代谢异常,可有心律失常[2]。本文总结了1例CGL1型伴发疹性黄瘤患者的临床特点及基因型,为临床和基因诊断该病提供依据。
1 材料与方法
1.1 对象
北京协和医院内分泌科2023年7月19日收住入院的一例14岁1个月的女性患儿,以“口干、多饮、皮下脂肪减少伴散在皮疹”为主要临床表现,入院后完善患儿的病史采集、体格检查及辅助检查,符合CGL诊断。为进一步明确该病的基因突变类型,抽取患儿外周血,行全外显子测序分析及 Sanger 测序验证,本研究获得了北京协和医院伦理委员会的批准(伦理审批文号: K2917),患儿监护人签署了知情同意书。
1.2 方法
1.2.1 实验室检查:对患者进行了血常规、尿常规、肝肾脂全、凝血功能等常规检测,完善了空腹及餐后2 h血糖、胰岛素、C肽水平检测,同时完善了骨代谢、补体2项、免疫球蛋白3项、1型糖尿病相关自身抗体谱(4项)、抗核抗体谱(17项)、类固醇激素全套等检查。
1.2.2 彩色超声检查:对患者进行了乳腺及腋窝淋巴结、颈动脉、锁骨下动脉、上肢动脉、椎动脉、肾动脉、外周血管动脉、下肢动脉、子宫双附件以及双肾、输尿管、膀胱彩色超声检查。
1.2.3 基因检测:由北京贝瑞和康医学检验实验室有限公司完成,抽取患儿外周静脉抗凝血4 mL,使用全外显子组高通量测序检测技术,利用Verita Trekker®变异位点检测系统(贝瑞基因)和Enliven®变异位点注释解读系统对数据进行分析,根据目标序列捕获高通量测序的结果设计引物,进行目标序列的PCR扩增及Sanger测序。
1.2.4 其余检查:包括眼底检查、肌电图、骨龄相(手、肘)、超声心动图及全身核磁共振成像检查。
2 结果
2.1 临床资料
患者为14岁1个月的女性,第2胎第2产,母亲孕期平顺(母高龄产妇,生产时40岁),足月顺产(出生体重4.1 kg,身长60 cm,Apgar评分10分),自幼体型消瘦,食欲及进食正常,走路、出牙、说话年龄较同龄儿无异,学习成绩好,不易生病。12岁1月时无诱因出现口干,多饮(2 L/d),尿量增加(具体不详),未予诊治,随后症状逐渐加重,偶发腹痛(全腹疼痛,1~2次/月,性质不易描述),间断出现恶心、呕吐、乏力。13岁2个月时患者发现血糖及血脂增高,应用胰岛素超过100 U,血糖控制仍不佳,无酮症酸中毒倾向。1年前双手肘伸侧逐渐出现散发皮疹,高于皮面,否认红肿、压痛、瘙痒,后蔓延至四肢,轻微剐蹭后可破溃、结痴,病理提示黄色瘤。病程中否认胰腺外伤及手术史,否认听力受损、面容改变、手足增大、脸变红圆、皮肤紫纹,否认怕热、手抖、多食、易饥,否认阵发性头痛、视物模糊、手足麻木、直立性低血压。患者精神可,睡眠可,食欲好,主食5~6两/d,肉2~3两/d,蔬菜少量,基本无运动。平素大便1次/d,为黄色成形软便,有持续的泡沫尿,夜尿2-3次/晚,近1年体质量无明显改变。既往史、个人史无特殊;目前月经未来潮;家族史方面,母亲及两个舅舅有糖尿病,否认高脂血症,父母非近亲结婚。
2.2 体格检查
体温为36.2 ℃,心率为121次/min,呼吸频率为18次/min,血压为120/79 mmHg(1 mmHg=0.133 kPa), 血氧为97%,身高为162 cm,体质量为46 kg,BMI 17.52 kg/m2,腰围67 cm,卧位血压120/79 mmHg,心率121次/min;立位血压123/78 mmHg,心率131次/min。查体发现,发育正常,营养良好,神清语利。细小尖脸,体型消瘦,全身皮肤脂肪菲薄,未见黑棘皮征,四肢静脉显露,肌肉显间,臀部凹陷。肩部、肘部及手臂、双膝关节及腿部散在凸起于皮肤表面的黄色丘疹,部分可见破溃、结痂。查体未见锁骨上脂肪垫、水牛背。心肺腹查体无殊,双乳Ⅴ期,阴毛Ⅰ期,双下肢不肿。
2.3 辅助检查
实验室检查包括:1)全血细胞分析:血小板为315×109/L,白细胞为7.37×109/L,中性粒细胞百分比为48.2%,血红蛋白为144 g/L。尿常规:尿糖≥55 mmol/L,尿酮体为1.5 mmol/L。凝血2:凝血酶原时间为10.2 s,凝血酶原活动度为122.9%,纤维蛋白原为 4.57 g/L,D-二聚体为0.15 mg/L。2)肝肾功能及血脂水平:白蛋白为49 g/L,胆固醇为9.45 mmol/L,三酰甘油为37.61 mmol/L,高密度脂蛋白胆固醇为0.56 mmol/L,低密度脂蛋白胆固醇为1.48 mnol/L,肌酐为30 μmol/L,血电解质正常;3)空腹血糖为14.1 mmol/L,空腹胰岛素为10.4 μIU/mL、C肽为1.40 ng/mL,餐后2 h血糖为16.8 mmol/L,餐后2 h胰岛素为 33.1 μIU/Ml、C肽为1.95 ng/Ml,糖化血红蛋白为13.4%;4)生长激素为1.3 ng/mL,胰岛素样生长因子1为 227 ng/mL;5)补体2项+免疫球蛋白3项:补体 C3为 1.322 g/L,补体C4为 0.254 g/L,免疫球蛋白G 5.80 g/L,免疫球蛋白A为1.27 g/L,免疫球蛋白M为0.62 g/L;6)1型糖尿病相关自身抗体谱(4项):胰岛素抗体阴性,胰岛β细胞抗体阳性,39.11 RU/mL,谷氨酸脱羧酶抗体阴性;7)抗核抗体谱(17项)阴性。
彩色超声检查包括:1)心脏:心脏射血分数为74%,心脏结构功能未见异常;2)肝胆胰脾超声显示轻度脂肪肝、肝大、脾大;3)乳腺及腋窝淋巴结;4)颈动脉等血管彩超均未见明显异常;5)子宫双附件彩超显示双卵巢体积大,左卵巢4.3 cm×2.9 cm×1.7 cm,内见多个卵泡样回声,较大者直径0.5 cm,右卵巢4.2 cm×3.1 cm×1.8 cm,内见多个卵泡样回声,较大者直径0.6 cm;6)双肾、输尿管、膀胱彩超发现双肾体积大,右肾长径13.3 cm,左肾长径13.6 cm,双肾皮质回声稍增强。
其余检查包括骨龄相(手、肘),阅片骨龄大小为15-16岁;全身核磁共振成像显示周身脂肪减少;眼底检查未见糖尿病眼底改变。
2.4 基因检测分析
本例患儿的Sanger测序法检测结果(图1)显示:受检者在基因AGPAT2上发生c.202C>T和c.646A>T两处杂合突变,无其他亲属验证,突变来源未知。该患儿第9号染色体上的AGPAT2基因存在第2外显子c.202C>T:p.R68*杂合无义突变和第5外显子的c.646A>T:p.K216*杂合无义突变。根据美国医学遗传学与基因组学学会(ACMG)指南及指南应用建议,提示基因AGPAT2的c.202C>T突变为致病突变位点,基因AGPAT2的c.646A>T突变为可能致病突变位点。基因AGPAT2的c.202C>T突变使对应密码子突变为终止密码子,导致蛋白长度截短,可能引起功能发生改变。基因AGPAT2的c.646A>T突变发生于基因的倒数第二个外显子的最后50个碱基,预测不会发生无义突变介导的mRNA降解,超过10%的蛋白区域受影响。
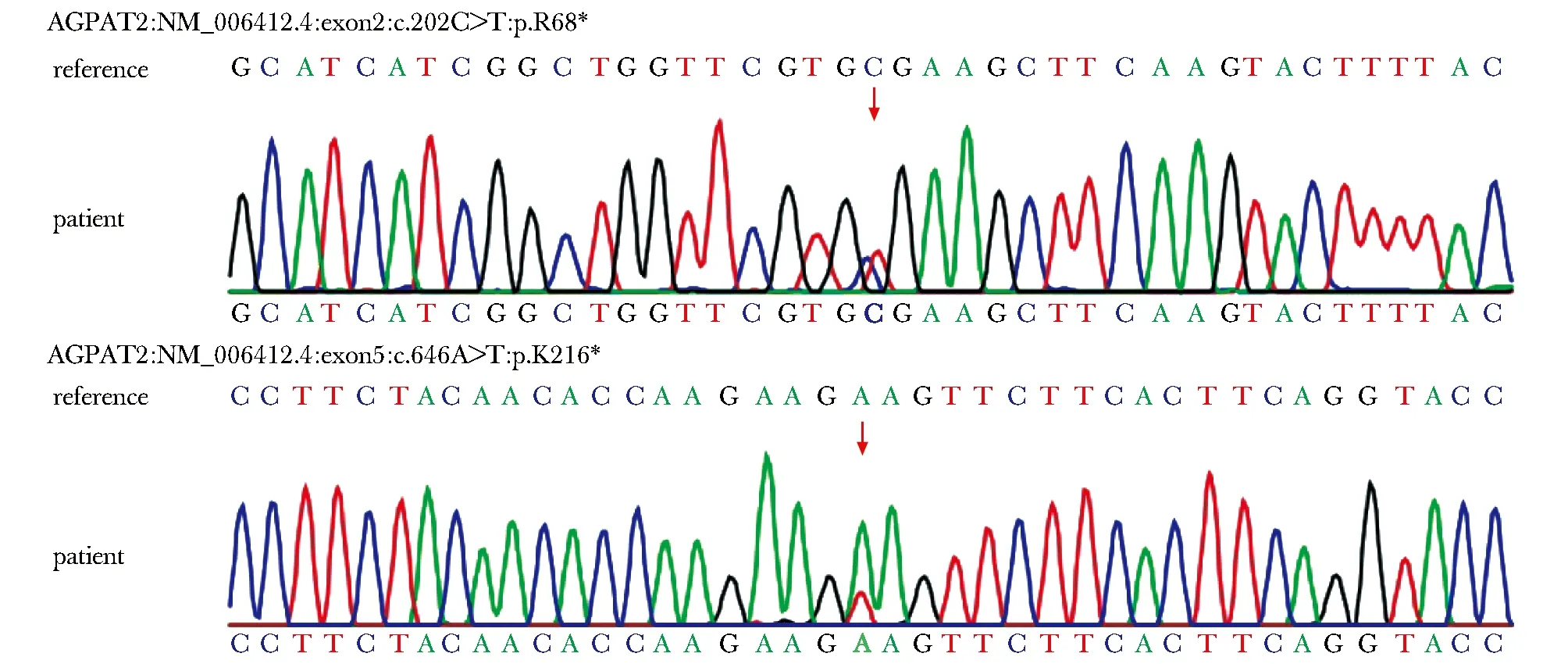
图1 本例患儿AGPAT2基因杂合突变Fig 1 Patient’s heterozygous mutation of AGPAT2 gene
2.5 诊断及治疗
根据本例患者的基因检测结果,结合临床表现,患者诊断为CGL1型明确。治疗方面,尚无有效的根治方法,主要是生活方式干预以及根据具体症状对症处理。患者应摄入血糖生成指数≤55的食品,低脂饮食,适度运动,注意休息。糖尿病方面,由于该患者存在严重的胰岛素抵抗,在胰岛素治疗同时可联用胰岛素增敏剂,首选二甲双胍,鉴于以往胰岛素治疗降糖仍不达标,可考虑加用恩格列净,同时对心、肾有保护效果。嘱规律应用地特胰岛素12 U~16 U早餐时及睡前皮下注射,门冬胰岛素16U-16U-16U三餐前联合格华止0.5 g/次,每日4次、恩格列净10 mg/次,每日1次治疗。高脂血症方面,加用非诺贝特200 mg,每晚睡前服用,协助调脂治疗,同时监测血脂水平。皮疹方面,考虑与血脂异常相关,积极降脂治疗后好转,皮损处对症外用复方多黏菌素B 外用,每日2次,避免外伤。窦性心动过速方面,可服用倍他乐克25 mg/次,每日2次,监测心率及心电图,建议避免大强度体力活动。
3 讨论
AGPAT2基因位于9号染色体,编码的蛋白由278个氨基酸组成,属酰基转移酶家族成员,在脂肪组织中高表达[1]。AGPAT2蛋白具有催化溶血磷脂酸的酰化形成磷脂酸的功能,在生物合成三酰甘油中起关键作用[1]。AGPAT2基因突变可导致脂肪细胞三酰甘油生物合成和储存障碍、脂肪细胞凋亡加速进而导致脂肪营养不良,CGL1患者可表现出脂肪萎缩、高三酰甘油血症、胰岛素抵抗等[4]。
以关键词“脂肪营养不良”“lipodystrophy”“CGL1”“AGPAT2”,分别在中国知网、万方、Pubmed数据库进行检索,检索时间截至2023年9月28日,共检索到69篇、88篇、116篇文献,通过逐篇阅读筛选,发现AGPAT2突变的CGL1病例目前在国内仅报道了6例[1-2,4-7](本例是第7例),均为近4年内报道。所有患者均有脂肪萎缩、肌肉明显的表现,除1例患者[4]外,其余病例[1-2,5-7]均发现有肝肿大表现(包括本例)。除3例患者[4,6- 7]外,其余病例[1- 2, 5]都合并有糖尿病。本例受检者AGPAT2基因的c.646A>T突变在其余2例[4-5]病例中也有报道,而本例报道的c.202C>T突变目前未有国内病例报道。c.646A>T突变属于无义突变,可导致终止密码子的提前出现,但可能因其突变位置位于基因编码区尾端(基因编码区3’端),且本例中为非纯合突变,推测可能保留了部分酶活性,从而造成一些临床表现型相对较和缓。
以关键词“脂肪营养不良”“lipodystrophy”“CGL1”“AGPAT2”“发疹性黄瘤”“ eruptive xanthoma”,分别在中国知网、万方、Pubmed数据库进行检索,检索时间截至2023年9月28日,未检索到文献。所以目前国内CGL1病例尚未有合并发疹性黄瘤的报道。
黄瘤病是一类常分布于皮肤或肌腱的黄色、棕红色的斑片、丘疹或结节,根据部位及形态的不同,可分为结节性黄瘤、发疹性黄瘤等[8]。发疹性黄瘤皮损常见于臀部、肩部和四肢伸侧,常见病因是高脂血症,也可继发于肥胖、糖尿病等,提示机体存在潜在的脂代谢紊乱[9]。一般黄瘤本身无需特殊处理,通常会随血脂的控制而减轻或消退。
本研究存在一定的局限性,缺少对患者家系进行AGPAT2基因突变位点的检测,拟计划在患者后续随访中完善其父母的基因检测,进一步确定患者AGPAT2基因的突变来源。本研究未对突变基因进行功能验证,虽然软件能够预测位点突变的致病性,但仍然不能取代功能试验。
综上,对临床表现为脂肪萎缩、胰岛素抵抗、高三酰甘油血症且合并发疹性黄瘤的患者要考虑有CGL的可能,建议完善生化检查,并行基因检测进一步精准诊断。
