光臀八齿小蠹共生微生物群落组成及其功能基因注释
2023-11-30刘彩霞梁玲瑜韩富忠王慧敏
刘彩霞, 李 刚, 梁玲瑜, 韩富忠, 王 正, 王慧敏, 吕 全,*
(1. 中国林业科学研究院森林生态环境与自然保护研究所, 国家林业和草原局森林保护学重点实验室, 北京 100091;2. 内蒙古自治区巴彦淖尔市乌拉特前旗森林病虫害防治检疫站, 巴彦淖尔 015006;3. 青海省黄南州麦秀林场, 黄南 811300; 4. 山东农业大学植物保护学院, 泰安 271018)
昆虫共生微生物是一个较为庞大的生物群体,包含细菌、真菌、古菌、病毒及小型原生生物(王渭霞等, 2021; 张娜等, 2022)。共生微生物为昆虫的生长、发育和繁殖等提供营养资源(Klepzig and Wilkens, 1997; Guevara-Rozoetal., 2020),在调节昆虫宿主化学信息交流与新陈代谢(Zhaoetal., 2019b),帮助昆虫解毒有害物质(Waterworthetal., 2020; Xuetal., 2022),保护其免受病原物危害(Zhouetal., 2016; Rangeretal., 2018)等方面有不可或缺的作用。小蠹虫作为针叶林中危害较为严重的害虫之一,与真菌和细菌等微生物形成了密切的共生关系(Linnakoskietal., 2012)。该类害虫可通过体外共生的模式,如体表的特殊结构(贮菌器) (Klepzig and Wilkens, 1997; Wangetal., 2013)或凹陷处(Kostovciketal., 2015)携带和传播共生菌。如由微囊目(Microascales)和长喙壳目(Ophiostomatales)组成的长喙壳类真菌是小蠹虫体表最主要的类群之一,该类真菌为小蠹虫整个生活史提供发育所需的碳源、氮源和甾醇等营养资源(Guevara-Rozoetal., 2020),也在防御性化合物等有害物质降解方面起重要作用(DiGuistinietal., 2011; Vanderpooletal., 2018)。除了上述体外共生菌,昆虫肠道共生菌也可以协助宿主获得必需的营养物质,从而影响小蠹虫的生长发育,如肠道分离出的酵母可以为根大小蠹Dendroctonusrhizophagus发育提供食物来源(Briones-Robleroetal., 2017),还有一些肠道共生微生物可以产生降解萜烯类(Adamsetal., 2013)、酚类(Zhaoetal., 2019a)、生物碱(Ceja-Navarroetal., 2015)、纤维素和半纤维素(Briones-Robleroetal., 2017)等相关酶来解毒植物防御性化合物。因此,体表和肠道微生物均在小蠹虫生长发育以及定殖过程发挥着不可或缺的作用。
目前,基于传统的分离培养、联合基因标记以及高通量测序技术对小蠹虫虫体、坑道和肠道共生微生物群落结构与组成及物种多样性进行了广泛的研究(Changetal., 2019; Chakrabortyetal., 2020a; Wangetal., 2020, 2021)。由于自然界约80%~90%的微生物无法利用传统方法进行分离培养(MsangoSokoetal., 2020),特别是与小蠹虫长期协同进化形成共生关系的微生物类群绝大多数属于不可培养,而共生微生物的功能研究可为微生物间的相互作用以及二者协同进化提供深入认知。因此,研究非可培养微生物群落构成与功能是必不可少的(Hernández-Garcíaetal., 2017; 韩一多等, 2019; 王渭霞等, 2021)。目前,在中欧山松大小蠹Dendroctonusponderosae及坑道富含有降解萜烯类化合物相关的功能细菌假单胞杆菌属Pseudomonas(Adamsetal., 2013)、咖啡果小蠹Hypothenemushampei降解咖啡因机制的功能菌假单胞菌目(Pseudomonadales)、肠杆菌目(Enterobacteriales)、根瘤菌目(Rhizobiales) (Ceja-Navarroetal., 2015; Vegaetal., 2021)、红脂大小蠹Dendroctonusvalens营养物质降解的功能细菌沙雷氏菌属Serratia和欧文氏菌属Erwinia(Zhouetal., 2016; Liuetal., 2022)以及降解淀粉、酯酶类和木聚糖营养的根大小蠹D.rhizophagus功能菌假单胞菌属、拉恩氏菌属Rahnella和假丝酵母属Candida(Briones-Robleroetal., 2017)等展开研究,为深入探究小蠹虫对营养资源的利用、萜类等防御性有毒物质的降解机制以及共生微生物间的互作提供潜在资源。目前,小蠹虫关键微生物功能已有一定的研究,但还没有详细的阐明(Hernández-Garcíaetal., 2017; Mayersetal., 2020)。
光臀八齿小蠹Ipsnitidus隶属于鞘翅目(Coleoptera)象甲科(Curculionidae)小蠹亚科(Scolytinae)齿小蠹属Ips,是中国特有物种,仅分布于中国青藏高原及其周边的高海拔地区(Jakušetal., 2010; 王正等, 2021)。该害虫主要危害青海云杉Piceacrassifolia和川西云杉Picealikiangensisvar.balfouriana的风倒木、衰弱木和濒死木等,大暴发时可危害健康树木,严重影响该地区森林生态系统稳定和工业木材的生产(Liuetal., 2008; 薛永贵, 2008; Jakušetal., 2010)。目前尚未见光臀八齿小蠹共生微生物群落组成和功能的报道。昆虫共生微生物的研究,目前多数工作是分别对肠道和体表微生物群落开展的,很少有对二者进行直接比较分析。本研究采用宏基因组学方法对光臀八齿小蠹的肠道和其他组织共生真菌和细菌群落进行分析,旨在明确小蠹虫共生菌群落组成与多样性;通过比较肠道和其他组织共生真菌和细菌在不同分类水平和功能基因上的差异,以此确定肠道共生菌的物种和功能基因是否较其他组织更加丰富;筛选出与营养合成和萜烯类降解相关的功能基因以及相关酶并注释这些基因所在物种的分类地位,以此来预测这些相关基因的功能菌;通过肠道和其他组织共生真菌和细菌的相关性网络分析,以此来解析昆虫-共生微生物间复杂的共生关系,从而为森林病虫害防治提供科学基础。
1 材料与方法
1.1 供试昆虫
光臀八齿小蠹样本于2020年采自青海省黄南麦秀自然保护区(35°19′39″N, 101°56′10″E),将青海云杉同一树段剥取出的成虫放入透气的2 mL无菌离心管, 液氮速冻, 虫体共计210头。并于-80 ℃低温冰箱保存。上述光臀八齿小蠹样品利用奥林巴斯体视显微镜(型号: SZX16)对齿的数量、额瘤和翅盘进行形态鉴定(Cognato, 2015),并将虫体样本合并分为3等份(70头/份),于体视显微镜下将每头虫体的肠道取出。3份虫体的肠道样本分别编号为A1, A2和A3,除肠道以外的其他组织样本编号为B1, B2和B3,共计6个样本。用于提取总DNA以及后续的宏基因组测序等。

表1 光臀八齿小蠹共生微生物的宏基因组测序信息Table 1 Sequencing information of metagenomes of the symbiotic microbes in Ips nitidus
1.2 文库构建和高通量测序
将1.1节合并后的样本充分混匀,选取500 mg组织样本使用CTAB法提取总DNA。待DNA检测合格后用超声波机械打断,使DNA片段化,并进行纯化、末端修复、3′端加A、连接测序接头。用琼脂糖凝胶电泳进行片段大小选择,进行PCR扩增形成测序文库,建好文库先进行文库质检,质检合格(质量满足建库要求,总量满足2次及以上常规量建库)的文库采用Illumina NovaSeq6000(产地San Diego,试剂盒NovaSeq 6000 S4 Reagent Kit)进行上机测序(北京百迈客生物科技有限公司)。
1.3 数据分析
采用Fastp软件对原始读段进行质量控制并过滤,得到高质量测序数据。使用Bowtie2同宿主中欧山松大小蠹参考基因组(GenBank登录号: GCA_020466585.1)序列比对,去除宿主序列获得光臀八齿小蠹肠道和其他组织共生菌基因组测序有效读段。使用软件MEGAHIT进行宏基因组组装,过滤短于300 bp的contig序列(Lietal., 2015)。采用QUAST软件对组装结果进行评估(Gurevichetal., 2013)。采用MetaGeneMark软件Version 3.26 (http:∥exon.gatech.edu/meta_gmhmmp.cgi)的默认参数识别基因组中编码区域进行基因预测(Zhuetal., 2010)。利用MMseqs2软件Version 12-113e3 (https:∥github.com/soedinglab/mmseqs2)进行基因去冗余,相似性和覆盖度阈值分别为95%和90% (Steinegger and Söding, 2017)。该宏基因组数据的生物项目数据库号(BioProject ID)为PRJNA956988;生物样品号(Biosample ID)分别为SAMN34209768, SAMN34209769, SAMN34209770, SAMN34209771, SAMN34209772和SAMN34209773;序列片段归档号(Sequence ReadArchive, SRA)分别为SRR24210639, SRR24210638, SRR24210637, SRR24210636, SRR24210635和SRR24210634。
利用非冗余蛋白数据库(non-redundant protein sequence database, NR)对光臀八齿小蠹肠道和其他组织共生菌群落物种分类进行注释(邓泱泱等, 2006),并采用KEGG数据库对基因功能进行注释,以此筛选出真菌和细菌功能差异的基因以及与营养合成和萜烯解毒代谢相关的功能基因。基于百迈客平台(https:∥international.biocloud.net/)分析共生菌群落构成。基于R(version 3.6.3)中的igraph包或联川生物平台(https:∥www.omicstudio.cn/tool.)根据Spearman算法筛选正相关性阈值≥0.5,负相关性阈值≤-0.5,且P<0.05,物种数80的数据组进行相关性网络数据分析。宏基因组的基因相对丰度的计算公式(Qinetal., 2012)如下:
ai表示i基因的相对丰度;Li和Lj表示i和j基因的长度;xi和xj表示i和j基因出现的次数;n表示总基因的数量。
2 结果
2.1 测序数据质控
结果显示,光臀八齿小蠹3份肠道样本和3份其他组织样本共6个样品的测序量呈上升趋势,最后渐进平缓,表明光臀八齿小蠹共生微生物基因组测序深度充足。在去除宿主基因组后共生微生物宏基因组组装的大小范围为15.84~18.08 Mb,注释到的共生微生物基因数目为111 009~157 486个,平均每个样本的基因数目为130 213个,测序与组装的百分比在96.10%以上,说明光臀八齿小蠹共生微生物宏基因组数据的质量较高(表1)。
2.2 共生微生物群落组成
比对NR数据库鉴定到3 520种共生微生物,隶属于细菌界(Bacteria)、真菌界(Fungi)、古菌界(Archaea)、病毒界(Viruses)等。细菌和真菌的多样性最高,分别占所有共生微生物物种数的69.01%和23.21%。尽管未匹配和未分类物种多样性较低,但平均相对丰度达79.93%;其次为细菌和真菌的平均相对丰度,分别为13.79%和9.69%。毛霉门(Mucoromycota)、子囊菌门(Ascomycota)、担子菌门(Basidiomycota)、壶菌门(Chytridiomycota)和捕虫霉门(Zoopagomycota)为主要真菌优势门,在其他组织中的相对丰度较肠道更高;变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、软壁菌门(Tenericutes)和厚壁菌门(Firmicutes)是细菌中的优势菌门,仅放线菌门的相对丰度在肠道较其他组织中高(表2)。
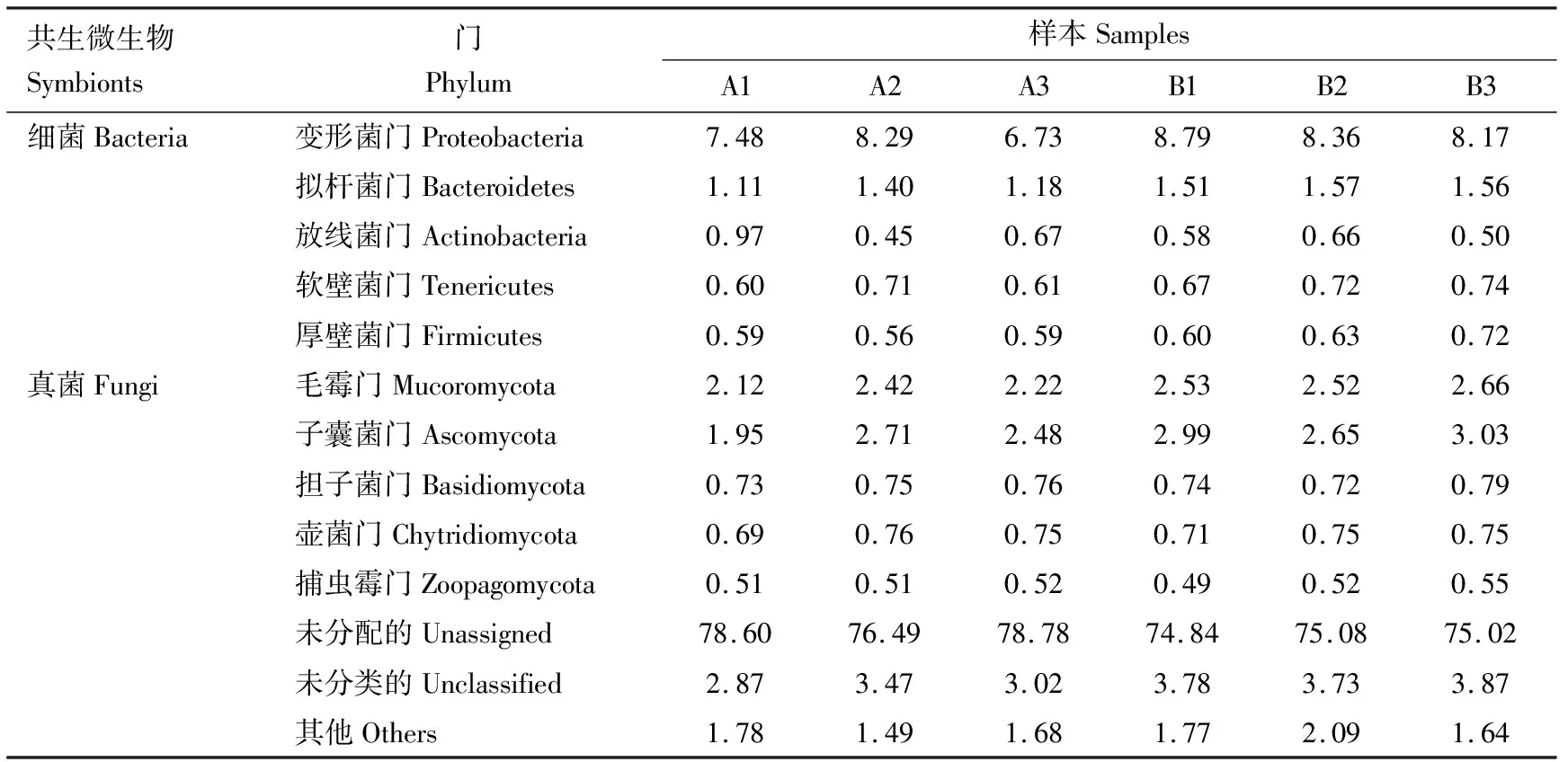
表2 光臀八齿小蠹共生微生物门水平的相对丰度(%)Table 2 Relative abundance (%) of the symbiotic microbes at the phylum level in Ips nitidus
共鉴定到共生真菌9门40纲93目217科403属743种,未分类74个(表3)。子囊菌门多样性和平均相对丰度均最高,该类真菌多样性和平均相对丰度分别为61.20%和32.06%(图1: A和表3)。多样性较高的菌门依次为担子菌门、毛霉门和微孢子门(Microsporidia)(表3),其平均相对丰度占整个真菌平均相对丰度的比例依次为9.09%, 29.34%和3.93%,且在其他组织中较在肠道中的相对丰度更高(图1: A)。子囊菌门和毛霉门均都占所有共生真菌平均相对丰度的60%以上。芽枝霉门(Blastocladiomycota)和隐真菌门(Cryptomycota)在肠道中的相对丰度高于在其他组织中的;从子囊菌目水平来看,白粉菌目(Erysiphales)、酵母菌目(Saccharomycetales)、散囊菌目(Eurotiales)和肉座菌目(Hypocreales)为真菌优势目,酵母菌目在肠道中的相对丰度略高于在其他组织中的,平均相对丰度占子囊菌门真菌的9.27%,而白粉菌目平均相对丰度最高,占子囊菌的63.66%(图1: B)。白粉菌属Erysiphe、丛枝菌根属Rhizophagus、蛙粪霉属Basidiobolus、根霉属Rhizopus、Spizellomyces、罗兹菌属Rozella、蛙壶菌属Batrachochytrium、被孢霉菌属Mortierella、横梗霉属Lichtheimia和柄锈菌属Puccinia等是相对丰度较高的优势真菌属。白粉菌属和丛枝菌根属均是肠道和其他组织的核心属。共注释到长喙壳类真菌微囊目和长喙壳目6属16种(参考物种),在其他组织和肠道中的平均相对丰度分别为0.016%和0.013%,其中,Grosmannia1种、Ophiostoma5种、Sporothrix3种、Ceratocystis3种、Leptographium2种、Esteya1种和1个未分类物种,在其他组织中的相对丰度较在肠道中的更高,Esteya仅出现在他组织中(图1: C)。
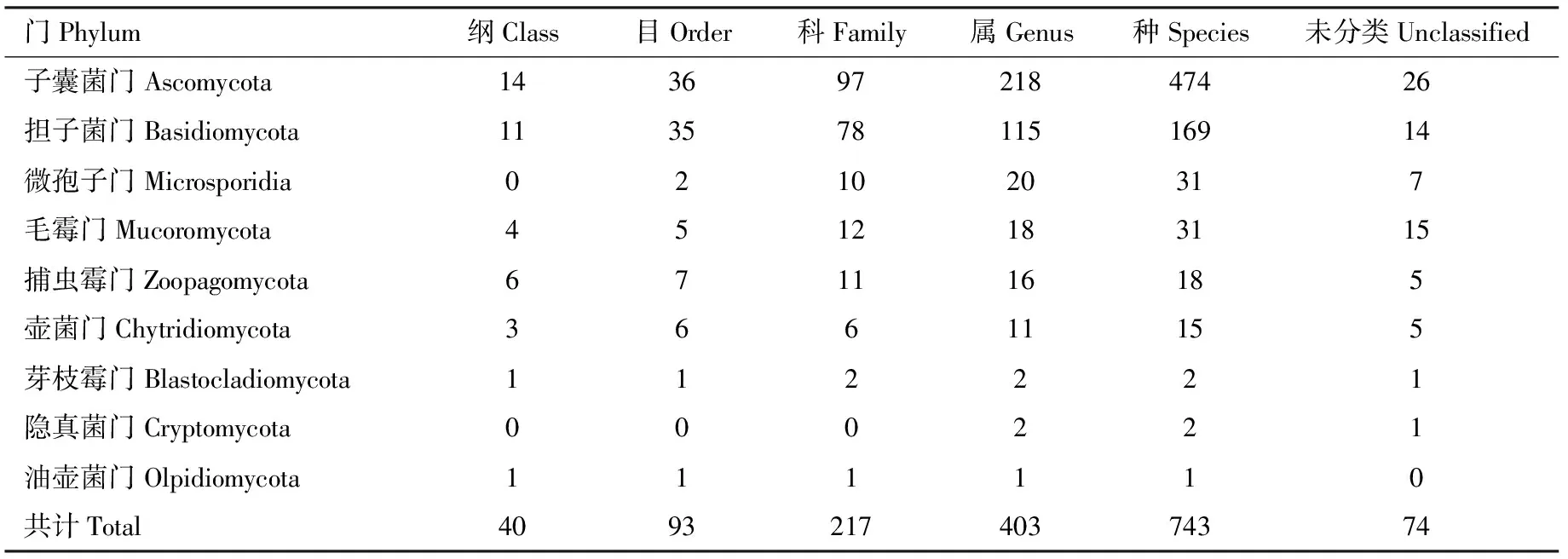
表3 不同分类水平下光臀八齿小蠹共生真菌群落Table 3 Symbiotic fungal communities at different classification levels in Ips nitidus
共鉴定到共生细菌35门69纲142目308科826属2 275种,未分类154个(表4)。变形菌门是优势菌门,其物种多样性和平均相对丰度约分别占整个共生细菌的45.82%和57.80%,其在其他组织中的相对丰度较在肠道中的更高,厚壁菌门、拟杆菌门和放线菌门多样性较高,其平均相对丰度占整个细菌群落的比例分别为4.46%, 10.06%和4.64%,放线菌门在肠道中的相对丰度高于在其他组织中的(图2: A)。γ-变形菌纲(γ-Proteobacteria)、α-变形杆菌纲(α-Proteobacteria)、β-变形菌纲(β-Proteobacteria)、鞘脂杆菌纲 (Sphingobacteriia)在其他组织中相对丰度较肠道中的相对丰度更高。肠杆菌目(Enterobacterales)、鞘脂杆菌目(Sphingobacteriales)和立克次体目(Rickettsiales)相对丰度较高,分别为23.53%, 6.79%和6.40%,在其他组织中相对丰度较肠道中的更高(图2: B)。链霉菌属Streptomyces、芽孢杆菌属Bacillus、假单胞杆菌Pseudomonas和泛菌属Pantoea的多样性较高,分别占细菌群落多样性的1.81%, 1.56%, 1.40%和1.32%,其中链霉菌属和泛菌属在其他组织中的相对丰度较肠道中的更高;芽孢杆菌属和假单胞杆菌属在肠道相对丰度较在其他组织中的更高。肠杆菌属Enterobacter、鱼立克次体属Piscirickettsia、沃尔巴克氏体属Wolbachia的相对丰度依次较高,分别为18.93%, 3.01%和2.71%,在其他组织中的相对丰度较肠道中的更高,且肠杆菌属和沃尔巴克氏体属均为肠道和其他组织的核心属(图2: C)。

图2 光臀八齿小蠹共生细菌在门(A)、目(B)和属(C)水平的相对丰度Fig. 2 Relative abundance of symbiotic bacteria in Ips nitidus at the levels of phylum (A), order (B) and genus (C)
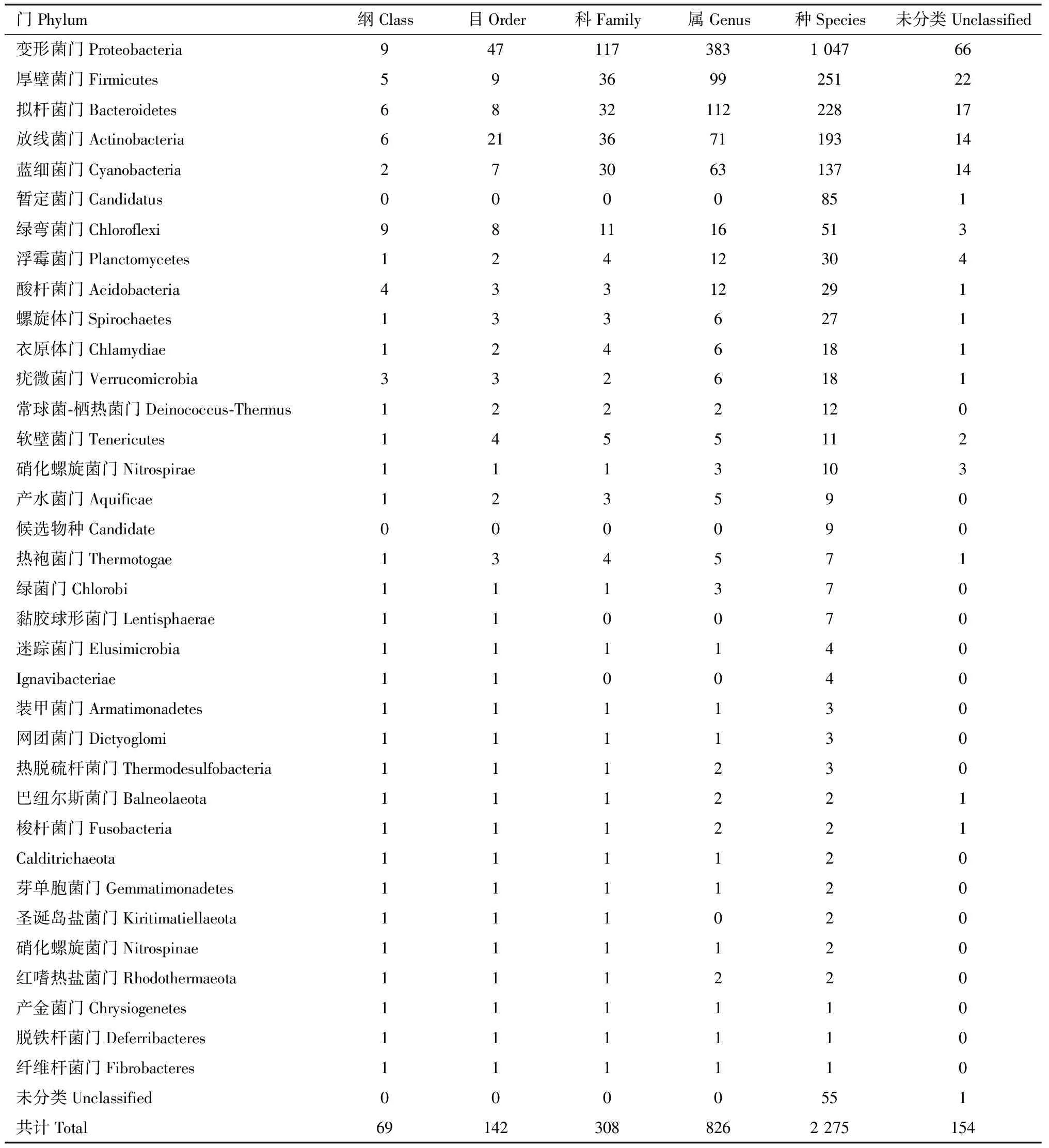
表4 不同分类水平下光臀八齿小蠹共生细菌群落Table 4 Symbiotic bacterial communities at different classification levels in Ips nitidus
2.3 共生微生物的相关性网络
网络分析显示,在肠道和其他组织中共生真菌和细菌间的正相关性P(互惠共生或共栖)均大于负相关性N(拮抗或抑制作用)(肠道:P=69,N=31; 其他组织:P=64,N=36),证明光臀八齿小蠹共生菌间存在较强的互惠共生或共栖关系。而且肠道和其他组织中在属水平相互作用的真菌的数量高于细菌的数量(肠道: 45属真菌,35属细菌;其他组织: 32属真菌,25属细菌),可能真菌和光臀八齿小蠹的共生关系较细菌和光臀八齿小蠹的共生关系更为紧密。在肠道,优势白粉菌属(肠道:P=34,N=9; 其他组织:P=12,N=9)和沃尔巴克氏菌属(肠道:P=18,N=5; 其他组织:P=7,N=5)的连接数高于其他组织中的,且正相关性高于负相关性,暗示了白粉菌属和沃尔巴克氏菌属在肠道比在其他组织中可能存在更为复杂的互惠共生相互作用(图3: A)。在其他组织中,丛枝菌根属(肠道:P=11,N=8; 其他组织:P=22,N=11)和肠杆菌属(肠道:P=6,N=9; 其他组织:P=23,N=11)的连接数均高于肠道的,表明这2个优势属在其他组织中较在肠道中存在更重要的相互作用(图3: B)。其他组织中优势肠杆菌属的正相关性高于负相关性(P=23,N=11),而在肠道表现相反(P=6,N=9)。一些优势菌属如海源菌属Idiomarina、产水菌属Aquifex、申氏杆菌属Shinella、假丝酵母属Candida和巨孢囊霉属Gigaspora等在肠道中呈现正相关性,在其他组织中呈现负相关性;不动杆菌属Acinetobacter、假单胞菌属Pseudomonas、微杆菌属Microbacterium、脱硫弧菌属Desulfovibrio、双珠霉属Dimargaris和内囊霉属Endogone在肠道中呈现负相关性,在其他组织中呈现正相关性。这些优势菌属与白粉菌属、丛枝菌根属和肠杆菌属相互联系,可能在光臀八齿小蠹共生菌间的相互作用有着重要的功能。

图3 光臀八齿小蠹肠道(A)和其他组织(B)中共生菌相关性网络Fig. 3 Correlation network of the symbionts in the gut (A) and other tissues (B) of Ips nitidus
2.4 共生菌功能基因注释
KEGG注释到了14 826个功能基因,包括共生真菌7 010个功能基因,细菌6 483个功能基因(图4)。这些功能基因均参与细胞过程(cellular processes)、环境信息处理(environmental information processing)、遗传信息处理(genetic information processing)和代谢(metabolism)。从真菌和细菌功能基因来看,真菌功能基因在KEGG一级水平遗传信息处理的全部通路以及细胞生长与死亡(cell growth and death)、运输和分解代谢(transport and catabolism)、信号传导(signal transduction)、多糖生物合成和代谢(glycan biosynthesis and metabolism)、核苷酸代谢(nucleotide metabolism)二级水平5个通路中较细菌基因丰富。真菌中没有注释到细胞过程中的细胞运动(cell motility)相关的功能基因,表明真菌可能没有参与到这一过程(图4: A)。细菌功能基因在全局和概述图谱(global and overview maps)、碳水化合物代谢(carbohydrate metabolism)、能量代谢(energy metabolism)和氨基酸代谢(amino acid metabolism)等数量高于真菌的,特别是与异源性物质生物降解和代谢(xenobiotics biodegradation and metabolism)相关的细菌功能基因明显高于真菌(真菌0.08%,细菌1.01%)(图4: B)。15条通路(代谢13条,遗传信息处理2条)在肠道和其他组织间细菌和真菌存在显著功能性差异。mRNA监测通路(mRNA surveillance pathway)、 丙酸代谢(propanoate metabolism)、泛素介导的蛋白质水解(ubiquitin-mediated proteolysis)、核黄素代谢(riboflavin metabolism)、嘧啶代谢(pyrimidine metabolism)、丙氨酸、天门冬氨酸和谷氨酸代谢(alanine, aspartate and glutamate metabolism)、N-聚糖的生物合成(N-glycan biosynthesis)、脂肪酸降解(fatty acid degradation)、酪氨酸代谢(tyrosine metabolism)、脂肪酸代谢(fatty acid metabolism)和多型N-聚糖生物合成(various types of N-glycan biosynthesis)在肠道较在其他组织的细菌和真菌基因的平均相对丰度高,表明可能光臀八齿小蠹共生菌这些通路在肠道较在其他组织的代谢作用更强(图4: C)。

图4 光臀八齿小蠹共生真菌(A)和细菌(B)的功能基因的KEGG通路以及肠道和其他组织间功能基因的差异KEGG通路(C)Fig. 4 KEGG pathways of the functional genes of the symbiotic fungi (A) and bacteria (B), and differential KEGG pathways of the functional genes between gut and other tissues (C) of Ips nitidus
KEGG注释到与异源性物质生物降解和代谢的细菌功能基因98个,真菌功能基因8个。参与到氯烷和氯烯降解(chloroalkane and chloroalkene degradation, ko00625)、苯甲酸酯降解(benzoate degradation, ko00362)、氨基苯甲酸酯降解(aminobenzoate degradation, ko00627)、己内酰胺降解(caprolactam degradation, ko00930)、萘降解(naphthalene degradation, ko00626)、苯乙烯降解(styrene degradation, ko00643)、氟苯甲酸酯降解(fluorobenzoate degradation, ko00364)、氯环己烷和氯苯降解(chlorocyclohexane and chlorobenzene degradation, ko00361)、硝基甲苯降解(nitrotoluene degradation, ko00633)、甲苯降解(toluene degradation, ko00623)、阿特拉津降解(atrazine degradation, ko00791)、类固醇降解(steroid degradation, ko00984)和二甲苯降解(xylene degradation, ko00622)13个通路,与细胞色素P450外源性物质代谢(metabolism of xenobiotics by cytochrome P450, ko00980)、细胞色素P450-药物代谢(drug metabolism-cytochrome P450, ko00982)以及药物代谢-其他酶(drug metabolism-other enzymes, ko00983)相关通路没有发现,猜测可能光臀八齿小蠹共生真菌和细菌不存在与细胞色素P450代谢的相关机制。氯烷和氯烯降解通路的功能基因最多(55个),其次为苯甲酸酯降解通路的功能基因(30个)。细菌基因主要分布于欧文氏菌属Erwinia、海源菌属Idiomarina、伯克霍尔德菌属Burkholderia、嗜盐单胞菌属Halomonas、假单胞菌属Pseudomonas、泛菌属Pantoea、沙雷氏菌属Serratia、巨型球菌属Macrococcus、芽胞杆菌属Bacillus等,真菌功能基因主要分布于球梳霉属Linderina、弹球菌属Sphaerobolus、异水霉属Allomyces等。
对于萜烯类降解的相关KEGG通路,仅注释到柠檬烯降解(limonene degradation, Ko00903)通路。6个真菌功能基因在肠道和其他组织中均有发现(表5),分布于Lobosporangium、Hesseltinella、巨孢囊霉属Gigaspora、Sugiyamaella、弹球菌属Sphaerobolus和异水霉属Allomyces。细菌功能基因57个,肠道细菌功能基因46个,其他组织细菌功能基因50个,主要分布于芽杆菌目巨型球菌属Macrococcus、海源菌属Idiomarina和假单胞菌属Pseudomonas等类群。此外,还发现甾醇合成 (steroid biosynthesis, Ko00100)通路,真菌功能基因11个,肠道中有12个,其他组织中有14个,主要分布于毛霉门根霉属Rhizopus和毛霉属Mucor、子囊菌门的裂殖酵母属Schizosaccharomyce和拟魏克酵母属Wickerhamomyces;担子菌门的串担革菌属Botryobasidium;芽枝霉门异水霉属Allomyces等以及分布于细菌变形菌门(Enhygromyxa)和甲基微菌属Methylomicrobium。
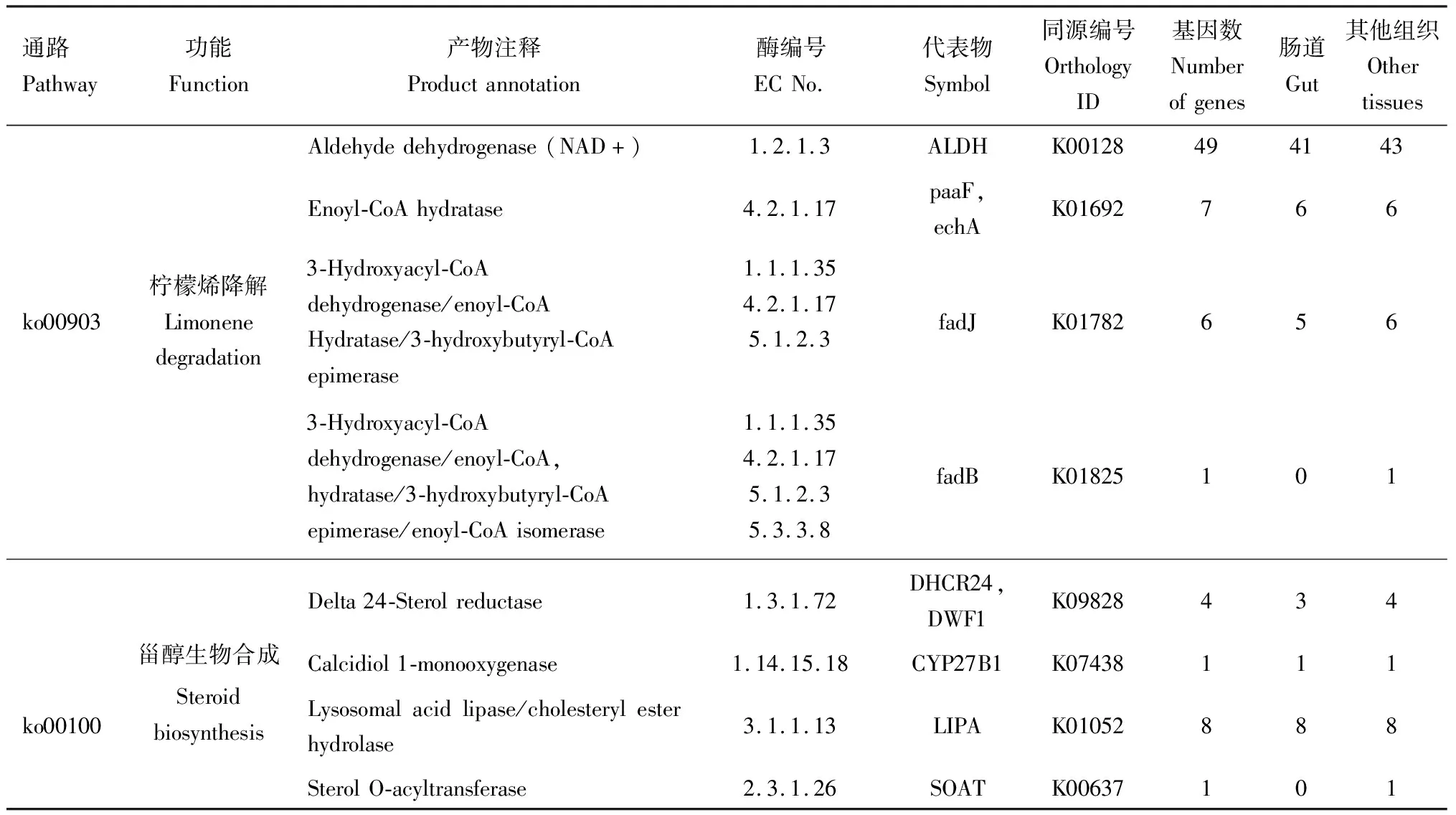
表5 注释到与单萜类柠檬烯降解和甾醇生物合成相关KEGG通路的光臀八齿小蠹共生真菌和细菌功能基因Table 5 Functional genes of the symbiotic fungi and bacteria in Ips nitidus annotated to KEGG pathways relatedto monoterpene limonene degradation and steroid biosynthesis
2.5 长喙壳类真菌相关功能基因
长喙壳类真菌6属16种共注释到功能基因74个,分布于Ophiostoma(肠道18个,其他组织27个)、Ceratocystis(肠道11个,其他组织15个)、Grosmannia(肠道8个,其他组织6个)、Sporothrix(肠道7个,其他组织9个)、Leptographium(肠道1个,其他组织2个)、Esteya(肠道0个,其他组织1个)和长喙壳科未分类种(ophiostomataceae unclassified)(肠道1个,其他组织2个)。这些基因主要参与到氧化磷酸化反应、代谢通路、氨基酸代谢、碳代谢等过程。Esteya仅在其他组织中被发现。碳代谢能力仅在Sporothrix、Ceratocystis和Grosmannia中发现,且Grosmannia在其他组织中较在肠道中的碳代谢能力更强,Leptographium参与到淀粉与蔗糖代谢,且在肠道中较在其他组织中的代谢能力更强。Sporothrix、Ophiostoma和1个长喙壳科未分类物种主要参与氧化磷酸化。
3 讨论
本研究采用宏基因组技术对光臀八齿小蠹肠道和其他组织的共生微生物群落组成与多样性以及功能基因进行分析,结果表明,未匹配和未分类物种的相对丰度最高,达79.93%,这可能与非可培养微生物无法利用传统方法分离获得有关,这与MsangoSoko等 (2020) 和Zhu等(2022)研究结果相一致。真菌中子囊菌门的物种多样性和相对丰度最高。光臀八齿小蠹肠道和其他组织中均有酵母菌目、肉座菌目、微囊目和散囊菌目等(图1: B)。该小蠹酵母目的物种多样性最高,约占全部真菌的12.21%。而白粉菌目的相对丰度最高,但在材小蠹Megaplatypusmutatus和华山松大小蠹Dendroctonusarmandi共生菌群落组成的研究中,发现酵母目的相对丰度最高(Huetal., 2015; Ceriani-Nakamurakareetal., 2018)。此外,担子菌门也是较为丰富的,这与Megaplatypusmutatus伴生真菌群落组成的研究相一致。白粉菌目在其他组织中较在肠道中表现出更高的相对丰度,而酵母目相反,即在肠道中较其他组织中具有更高的相对丰度。在长喙壳类真菌群落中,发现16种(参考物种),包含长喙壳目5属13种和微囊目1属3种。该类群的相对丰度较低,且在其他组织的相对丰度较肠道更高(图1: C),在其他齿小蠹类群中发现长喙壳科(Ophiostomataceae)和Ceratocystidaceae两个科是小蠹亚科肠道的核心菌(Chakrabortyetal., 2020b)。尽管该类群的相对丰度较低,但在小蠹虫营养来源(Guevara-Rozoetal., 2020)、化学信息交流(Zhaoetal., 2019a)、寄主防御性化合物降解(DiGuistinietal., 2011; Briones-Robleroetal., 2017)方面发挥着重要的作用。
从共生细菌来看,变形菌门、拟杆菌门、放线菌门和厚壁菌门等细菌群落的物种多样性和相对丰度均较高(图2: A)。先前对于中欧山松大小蠹(Adamsetal., 2013)和红脂大小蠹(Morales-Jiménezetal., 2009; Chengetal., 2018)的研究发现α-变形杆菌纲、β-变形菌纲、γ-变形菌纲和放线菌纲等存在于所有样本中,这与本研究的结果相一致。从属水平来看,光臀八齿小蠹共生细菌相对丰度较高的依次为肠杆菌属、鱼立克次体属、沃尔巴克氏体属、微粒孢子虫属Anaplasma和不动杆菌属Acinetobacter等,且其他组织中的相对丰度较肠道中的更高(图2: C)。而链霉菌属、芽孢杆菌属、假单胞菌属和泛菌属多样性较高,均存在于光臀八齿小蠹肠道和其他组织。而红脂大小蠹(Zhouetal., 2016)和根大小蠹等一些大小蠹物种(Briones-Robleroetal., 2017; Hernández-Garcíaetal., 2017; Ceriani-Nakamurakareetal., 2018)也存在这些细菌物种,并证明该类群与萜烯类、纤维类等防御性化合物的降解相关。但在本研究发现链霉菌属和泛菌属的相对丰度在其他组织中更高,而芽孢杆菌属和假单胞杆菌相对丰度在肠道较其他组织中更高。肠杆菌属作为共生细菌中一个丰度最高的类群,目前在咖啡果小蠹等其他小蠹物种(Hernández-Garcíaetal., 2017; Vegaetal., 2021)和山茶象Curculiochinensis等(Zhangetal., 2020)鞘翅目昆虫表现出较高的相对丰度,且为核心菌,该类群在解毒植物防御化合物过程中发挥着重要的作用。此外,肠杆菌属在鳞翅目的Samiaricini中也有发现,可以帮助调节肠道的pH而提高碱性环境,在单宁的降解中起重要的作用(MsangoSokoetal., 2020)。因此,肠道共生菌对昆虫的生长发育和宿主难利用物质降解有着重要的意义(Briones-Robleroetal., 2017)。而小蠹虫共生细菌群落组成可能受虫体本身的影响,也可能与虫体不同发育阶段相关,如红脂大小蠹肠道共生细菌Pseudomonasputida仅出现在的幼虫时期,Acinetobacterlwoffii可以在幼虫和羽化成虫前时存在,但Arthrobacteroxidans仅在幼嫩成虫时出现(Briones-Robleroetal., 2017)。与此同时,还可能因肠道分离部位以及采样地理区域不同从而导致肠道共生细菌群落组成和多样性存在差异(Chakrabortyetal., 2020b; Guevara-Rozoetal., 2020; MsangoSokoetal., 2020)。
此外,本研究分析了肠道和其他组织共生真菌和细菌群落间的相互作用。在肠道和其他组织共生真菌和细菌间的正相关相互作用(互惠共生或共栖)均高于负相关相互作用(拮抗或抑制)(图3: A和B)。表明小蠹虫共生菌间存在密切的互惠共生关系。而Zhu等(2022)在拟果蝇Drosophilasimulans和黄粉鹿角花金龟Dicranocephaluswallichiibowringi肠道真菌和细菌群落组装中,发现肠道微生物间可能存在更复杂的互惠共生作用。本研究还发现一些优势菌群海源菌属、产水菌属和申氏杆菌属等在肠道和其他组织中与白粉菌属、丛枝菌根属和肠杆菌属呈现相反的作用,如海源菌属、产水菌属、申氏杆菌属、假丝酵母属和巨孢囊霉属等在肠道呈现正相关作用,在其他组织中呈现负相关作用;不动杆菌属、假单胞菌属、微杆菌属、脱硫弧菌属、双珠霉属和内囊霉属在肠道呈现负相关性,在其他组织中呈现正相关作用。
在功能基因方面,发现光臀八齿小蠹共生真菌功能基因7 010个,细菌功能基因6 483个。真菌功能基因没有发现细胞运动(cell motility)相关的,在细菌功能基因中,发现异源性物质生物降解和代谢的基因明显高于真菌,主要分布于欧文氏菌属、海源菌属、伯克霍尔德菌属、嗜盐单胞菌属等类群。由于柠檬烯是一种重要的单萜类化合物,可以抑制小蠹和共生菌的存活。目前已有研究表明小蠹虫共生菌及其代谢产物能够协助宿主降解植物防御性化合物(DiGuistinietal, 2011; Ceja-Navarroetal., 2015; Xuetal., 2016)。本研究发现与萜烯类柠檬烯降解通路ko00903主要存在于巨型球菌属、海源菌属和假单胞菌属等类群。与此同时,发现的相关酶与Adams等(2013)对中欧山松大小蠹萜烯类降解的酶类相一致。但中欧山松大小蠹与萜烯类降解相关的功能基因主要分布于假单胞菌属和拉恩氏菌属等(Adamsetal., 2013),与本研究结果略有差异。
由于共生真菌可以为小蠹虫和寄主树木提供自身无法合成但生长发育必需的甾醇(Bentz and Six, 2006)。如中欧山松大小蠹可以直接从长喙壳类真菌共生体中获取麦角甾醇(Guevara-Rozoetal, 2020)。本研究发现与甾醇合成通路ko00100,其他组织和肠道的基因数分别为14个和12个(表5),表明其他组织较肠道可能为小蠹提供更多的营养来源。先前研究证明大小蠹的长喙壳类真菌中麦角甾醇是小蠹虫卵、幼虫和蛹生长发育必需的营养,但本研究未在长喙壳类真菌中发现。这些与萜烯降解和甾醇合成相关的异水霉属、海源菌属、假单胞菌属和毛霉属等是真菌和细菌群落的优势属,且与核心菌属丛枝菌根属、白粉菌属和肠杆菌属相互联系,表明相关性网络中的3个优势菌属可能在降解植物化合物和营养资源方面有着间接的作用。此外,DiGuistini等(2011)还发现长喙壳真菌Grosmanniaclavigera具有降解萜烯类有毒物质的基因。但在本研究中暂未在长喙壳类真菌发现可以降解寄主防御性化合物的功能基因。可能与小蠹虫共生菌群落特征和小蠹的分布有关相关(Bentz and Six, 2006; Guevara-Rozoetal., 2020)。但对于小蠹虫密切相关的伴生菌长喙壳类真菌Grosmannia、Ophiostoma和Ceratocystis的74个功能基因参与到氧化磷酸化、碳和能量代谢通路,且Ophiostoma是肠道和其他组织中功能基因最丰富的类群(Behmer and Nes, 2003)。
作为中国特有物种光臀八齿小蠹,该小蠹具有丰富的共生微生物,特别是细菌和真菌。共生子囊菌门的酵母菌和白粉菌是真菌的两个重要类群,分别在其他组织有着最高的物种多样性和相对丰度。在共生细菌群落组成中,证明变形菌门是其他组织中物种多样性和相对丰度最高的一个类群,特别是核心菌肠杆菌属,该类群是细菌群落中最为丰富的类群,且在其他组织的相对丰度较肠道更高,该类群在大小蠹等鞘翅目昆虫对于防御性化合物降解方面均有一定的研究,未来可对该类群的功能基因进行深入探讨,为研究防御性化合物降解提供新的思路。与此同时,小蠹虫共生菌在肠道和其他组织存在密切的互惠共生作用。此外,对于长喙壳类真菌的功能基因主要参与到了氧化磷酸化和氨基酸等代谢过程中。研究还发现单萜柠檬烯降解通路ko00903和甾醇生物合成通路ko00100。本研究为后续小蠹虫和共生菌间的共生机制和协同进化关系的深入探究提供基础数据。
