融合卷积与自注意力机制的基因型填补算法
2023-11-29陈炯环鲍胜利王啸飞李若凡
陈炯环,鲍胜利*,王啸飞,李若凡
融合卷积与自注意力机制的基因型填补算法
陈炯环1,2,鲍胜利1,2*,王啸飞1,2,李若凡1,2
(1.中国科学院 成都计算机应用研究所,成都 610213; 2.中国科学院大学,北京 100049)( ∗ 通信作者电子邮箱baoshengli@casit.com.cn)
基因型填补可以通过填补估算出在基因测序数据中未覆盖的样本区域弥补因技术限制导致的缺失,但现有的基于深度学习的填补方法不能有效捕捉到全序列位点间的连锁关系,造成整体填补准确率低、批量序列填补准确率分散等问题。针对这些问题提出一种融合卷积与自注意力机制的填补方法——FCSA,使用两种融合模块构成编解码器组建网络模型。编码器融合模块使用自注意力层得到全序列位点间的关联度,将该关联度融合到全局位点后再通过卷积层提取局部特征;解码器融合模块使用卷积对编码后的低维向量进行局部特征重建,应用自注意力层对全序列建模并融合。使用多物种的动物基因数据进行模型训练,并在Dog、Pig和Chicken数据集上进行比较验证,结果表明,与SCDA(Sparse Convolutional Denoising Autoencoders)、AGIC(Autoencoder Genome Imputation and Compression)和U-net相比,FCSA在10%、20%和30%缺失率下的平均填补准确率均取得了最高值,且批量序列填补准确率的分散程度较小;消融实验的结果也表明,这两种融合模块的设计能够有效提升基因型填补的准确率。
基因型填补;卷积;自注意力;融合模块;全序列建模
0 引言
全基因组关联分析(Genome-Wide Association Study, GWAS)能在全基因组范围内分析与识别出单核苷酸多态性(Single Nucleotide Polymorphism, SNP),并找出与疾病相关的位点[1]。进行GWAS这类下游任务需要使用位点覆盖率较高的基因数据,因此保证测序数据的覆盖率是一个重要因素。测序数据中未覆盖到区域的基因型未知,称这些区域的位点为缺失位点。因此,对包含缺失位点的非全测序数据进行基因型填补(genotype imputation)是一项重要的任务。
基因型填补是根据已观察到的基因型计算推断未进行基因分型的等位基因的预测方法[2]。基因型数据中的缺失很常见,导致这种缺失的因素很多,例如低检出率、Hardy‑Weinberg平衡的偏离以及大量的低频位点变异[3],除遗传性丢失外,仅因技术问题导致的检测性缺失就有多种。此外,随着基因芯片以及测序技术的发展,诸多商业化厂商针对动植物开发出不同的基因芯片,与全测序技术相比,使用基因芯片能极大地控制成本。现有基因芯片的测序能力涵盖低、中、高密度,以奶牛为例,主要使用中密度基因芯片,而高密度芯片测序及全测序技术由于价格昂贵不具有大规模使用的现实意义。应用基因型填补可以将较低密度芯片数据填充至较高密度甚至全测序数据,在不增加成本的情况下保证下游全基因组关联分析等任务的准确性[4-5],因此,有必要实现一种高效且准确的基因型填补方法。
基因型填补能够有效填补缺失位点的依据是基因之间存在连锁不平衡(Linkage Disequilibrium, LD),即几个位点在表达的过程中并非独立进行,某个位点的表达与它所在区域的位点密切相关,原因是基因序列中一定区域内的两个位点之间的等位基因在多代遗传中会出现非随机组合的现象,也正是因为存在这种关联关系,才可以去学习与捕获强弱关系从而进行填补[6]。现有的基因型填补技术有两种:一种方法是依靠许多样本的基因组参考面板,这些样本通常包含相同或相似的物种背景,通过计算并寻找现有面板数据中基因型之间的连锁关联性,从而对缺失基因进行填补。填补的方法主要包括基于隐马尔可夫模型(Hidden Markov Model, HMM)以及蒙特卡洛马尔可夫链(Markov Chain Monte Carlo, MCMC)等统计学的模型,与之对应的商用软件有IMPUTE系列[7]、MACH[8]、fastPHASE[9]等,它们各具优缺点,文献[10]中对此进行了详细介绍与总结。尽管传统方法能够完成这项任务,但由于HMM和MCMC模型本身的参数量不多,导致在面对海量基因样本时不能有效拟合,参数的迭代学习也导致填补时间较长,并且这类模型依赖参考面板,具有较强的独立性假设,很难挖掘出距离较长位点间的连锁关系,因此迫切需要一款针对大样本数据的填补准确率高且用时较短的方法模型[11]。另一种填补方法是在填补时不依靠基因组参考面板,而是通过模型对大量基因参数进行拟合,学习总体样本中基因的总特征,再根据具体特征对缺失部分进行拟合,这就要求模型具有较强的泛化能力。本文提出的应用深度学习的填补方法就是基于无参考面板的思路进行研究。
近年来,深度学习的突破推动了人工智能的再次发展,它的诸多模型方法可以起到强特征提取器的作用,能够很好地捕获基因位点之间的连锁关系。目前,将深度学习相关模型方法应用于基因型填补这一任务仍属于开创性工作,市面上还没有一款成熟的基于深度学习的填补工具。因此,本文提出一种融合卷积与自注意力机制的基因型填补方法FCSA(Fusing Convolution and Self-Attention),采用开源的动物测序数据作为支撑,将测序数据按照不同的比例随机缺失,构造缺失样本与非缺失样本并进行自监督学习,从而对序列进行建模。FCSA的模型中提出了两种融合模块:编码器融合模块与解码器融合模块,并通过这两种融合模块构建编解码器。其中,编码器融合模块能够通过多头自注意力(Multi-head Self-Attention, MSA)机制计算全序列的关联关系,融合到序列后再通过卷积层进行局部特征提取,使用该融合模块层编码,最终得到缺失基因序列的嵌入编码表示;解码器融合模块先提取低维编码的局部特征,再进行全序列的特征计算与融合,从而对编码后的缺失基因型进行更完整的特征重建。本文主要工作如下:
1)提出一种基于深度神经网络的无参考面板基因型填补方法FCSA,并通过实验验证该方法能够填补低密度测序,且填补准确率较现有基于无参考面板的深度学习方法均有提升。
2)对Transformer模块提出四点改进,设计了两种融合卷积与注意力机制的模块(编码器融合模块与解码器融合模块)构建模型,使用这两种模块构建的编解码器能够兼顾序列的全局与局部特征,更合理地进行特征提取与重建,有效捕捉不同距离位点之间的连锁关系。
3)验证模型在多物种基因数据上的填补能力,说明了该模型具有较强的鲁棒性,并通过消融实验验证了使用融合模块及模块中融合方式的有效性。
1 相关工作
目前,将深度学习应用于生物基因层面的工作较少,用于填补的相关工作仍处在一个探索的状态,SCDA(Sparse Convolutional Denoising Autoencoders)[12]使用了一种基于去噪自动编码器的填补方法,该方法使用一维卷积神经网络构建自动编码器,并证实了该方法在填补正确率上优于无参考面板的统计学模型;但卷积网络在层数较少的情况下难以对离散数据的全局特征进行有效的融合与建模,当缺失率较高时,模型的去噪能力略显不足。
受人工神经网络的启发,AGIC(Autoencoder Genome Imputation and Compression)[13]使用一种自动编码器的填补方法,模型采用6层前馈神经网络搭建自动编码器;但由于模型规模不大,参数量较少,输入网络的编码数据仅28 b,一次能填补的位点仅为个位数,填补效率极低。
近些年来,生成对抗网络在基因数据生成领域取得了不错的效果[14],但是,直接使用随机噪声生成该缺失样本的非缺失对象并不现实。与基因生成的思路相似,使用带有一定比例的缺失数据与完整的基因型数据联合训练生成器与判别器,以达到将带有缺失位点数据生成至完整数据的效果。但是该网络方法难以训练,甚至生成的序列与真实数据结构性差异较大。
基于图像重建的思想,文献[15]中提出了一种基于U‑net的基因型填补方法,采用全卷积网络和自动编码器还原数据的分布,将缺失的基因型数据通过编码器进行卷积和池化,再由解码器将编码后数据插值成目标尺寸,解码过程中融合相应编码层中的特征,完成对原始数据的重构工作。但是基因数据是离散型数据,相邻基因型存在的连锁关系与图像中连续像素点的关系并不同,难以通过图像重建的方式来捕获基因型关联,因此在正确率上也存在一定的欠缺。
2 FCSA模型
本文提出的FCSA模型包括编码器与解码器:编码器负责对输入的缺失基因型数据进行全局序列的关联性计算、局部关键特征的提取和融合,从而捕获序列之间的连锁关系;解码器用于编码后的特征数据重建与恢复,以此完成原始数据的重建工作。在编码器与解码器中都使用了融合卷积与自注意力机制的融合模块,这分别发挥了卷积神经网络强特征提取器以及自注意力机制对长距离数据建模的优势。
FCSA工作流程如图1所示:处理并读取用于基因填补的数据,在此过程中划分训练集、验证集、测试集;设置缺失比例并生成缺失位点索引矩阵,根据索引进行掩码。在输入模型前设置序列长度并划分数据,在设置批量大小后输入模型进行训练,最后通过计算训练损失、验证集损失、准确率、验证集准确率对模型进行评估并得到最优化模型。
2.1 问题定义
针对数据格式对问题进行定义,定义参考面板一共包含个位点,模型一次能处理的序列长度为个位点,那么参考面板中包含的批量总数如下:

在训练集中,完整的序列数据可以表示为:
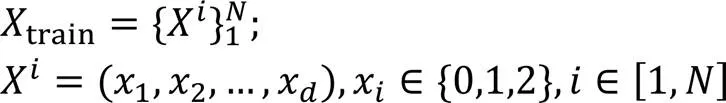
那么对应的模型输入的缺失样本可以表示为:

对样本进行编码后,假设每个位点数据对应的维度大小为,则每次流经模型的位点序列可以表示为:
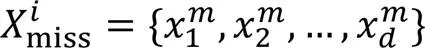

图1 FCSA工作流程
2.2 FCSA及其编解码器的构建
自动编码器(AutoEncoder, AE)是一个无监督的神经网络基础模型,旨在通过学习编码特征建模原始的数据表示,借助AE的思想,FCSA模型整体采用编解码器的网络结构,如图2所示,模型包括一个编码器、一个解码器。其中,编码器负责对输入的带有缺失的基因型数据进行特征提取,通过这个过程,模型可以提取输入数据的重要特征并表示到多个数据通道中去,得到一个压缩后的多通道特征向量; 编码器负责将特征提取后的元素进行重构与重新映射,通过最小化损失函数优化输入数据的重构,从而达到填补的目的。

图2 编解码器结构示意图
每一个带有缺失基因型数据流经模型得到填补后的完整基因数据的表示过程为:

整个FCSA模型由编码器与解码器构成,如图3所示。编码器的作用是对带有缺失位点的序列进行特征提取,得到原始序列中关键的连锁关系。在编码器中使用了三个Encoder Block融合模块,它们在结构上略有差异。由于第一个模块输入序列的维度较低,故采用2层3头自注意力,计算序列的全局关联度与输入序列各位点进行融合以突出关联特征。融合后的序列信息输入卷积网络有利于局部特征的提取,卷积操作能够在输入数据中捕捉到局部特征并生成一个更高维的特征映射,最大池化能够对卷积后的非重叠子区域进行最大值过滤,过滤出具有突出特征的位点。第二和第三个模块采用2层4头自注意力机制。三个融合模块中的卷积核的输出数量分别为32、64、128。

图3 FCSA的网络结构
解码器的作用是对编码器压缩后的特征向量进行重建,采用两个Decoder Block与一个卷积层。序列数据流经融合模块的一维卷积层后重构局部信息并降低维度,这个过程保留了原始位点的位置关系,注意力层计算全序列相似度并融合到位点序列,上采样层重构序列的关键信息。最后一层卷积重构序列维度至编码器输入的原始维度,并使用Softmax函数进行激活。解码器中所涉及的卷积层中卷积核的输出数量分别为64、32、3。
2.3 注意力机制
Transformer模型[16]目前已成为自然语言处理领域的主导模型,在处理较长序列时表现出了非常好的效果。自注意力机制是Transformer模型中基础的构建模块,它能够通过一定方式计算该序列中不同位置元素之间的相似度,将序列中的每个元素进行关联性建模,从而得到较长序列的表示。

通过与相乘得到相似度得分,经过Softmax处理后与进行加权,得到最终输出,整个过程可以表示为:

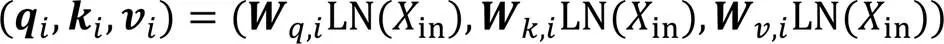
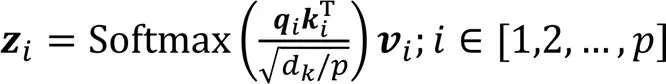
2.4 融合模块
卷积神经网络的优点是能够较好地对局部信息进行提取并保留位置信息,整体建模就需要增加卷积的层数,由于基因型数据是长序列,通过增加卷积层来扩大感受野的成本极高。注意力机制的优点是它能考虑到全时序信息,能够更有效地获得全局视野,但需额外引入并学习位置信息。因此,为了发挥两者的优势,对Transformer模块进行改进并提出两种融合模块,具体改进如下:
1)将Transformer原始模块中的前馈网络替换成一维卷积操作,这能更好地捕捉或重建局部特征;由于卷积操作可以学习到位点间的位置信息,因此不再需要位置编码。基因序列中基因型是离散的,定义离散空间中的卷积计算如下:


2)使用多层MSA网络,每层之间进行残差连接与层归一化,并在最后一层结束后融合输入序列,目的是增强局部关联性较强的位点特征,表示如下:

3)解码器融合模块重建出的序列要保留原始序列的位置信息,且编码后序列的整体感受野较小,因此在解码器融合模块中先采用一维卷积重建局部信息,再采用注意力层加强序列间的连锁关系,表示为:

4)在模块中融入特征维度压缩与重建的能力,编码器提取带有缺失的序列关系并逐步降维,解码器根据序列关系恢复至原始序列,因此在编解码器的最后添加下采样(Subsampling)与上采样(Upsampling)操作并配合Dropout来防止过拟合。
本文构建的Encoder Block与Decoder Block融合模块结构如图4所示。
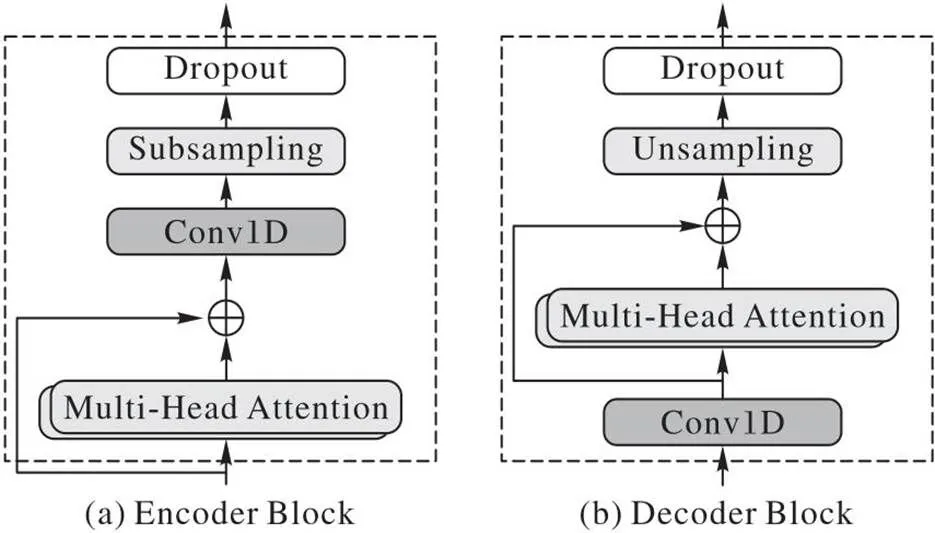
图4 编码器和解码器融合模块
2.5 损失函数
在构建FCSA整体结构后,由于序列中的位点信息是离散的,为了更准确地重构序列位点的对应编码,选择使用交叉熵损失函数来优化完整基因型序列与缺失序列的差距,公式如下:




3 实验与结果分析
3.1 数据集
本文使用的基因参考面板全部来源于Animal-ImputeDB的公共开源数据库[18],该数据库包含了13个动物物种的高质量基因组,致力于从各种研究和数据库中收集非人类动物的基因型数据和全基因组重测序数据。
本文实验采用的数据集为VCF格式的二倍体基因型数据。由于不同的物种数据具有不同的相关模式,位点模式的分散程度也会影响填补的准确率,为了消除数据对模型填补准确率的影响,本次实验选取采用三个物种的测序数据,分别为chr1_chicken、chr38_dog和chr4_pig。
在使用数据前首先要对VCF格式文件中的二倍体基因数据进行预处理,将0|0替换为0,1|0或0|1替换为1,将1|1替换为2,编码后的数据变为了一个由0、1、2组成的矩阵。
本文模型采用自监督的训练方式,设置缺失比例,通过编写脚本随机创建缺失位点并记录其索引,该脚本可以统计基因数据中的位点量,根据设置的缺失比例生成一个随机数组,通过随机数组可以将对应位置的基因型数据重置,从而达到设置位点随机缺失的效果。
根据问题性质,输入网络模型前对处理后的矩阵进行独热编码,将编码后的数据进行划分,训练集、验证集、测试集分别占70%、10%、20%。
3.2 实验环境及参数设置
本文对比实验、消融实验的训练及验证过程的服务器配置为:AMD EPYC 7302 CPU @2 GHz,NVIDIA GeForce RTX 3090 GPU;64 GB RAM;FCSA模型与对比实验模型全部基于Tensorflow 2.5.0、CUDA 11.2和Python 3.8环境完成。
模型的batch size设置为64,采用Adam优化器,学习率设置为0.000 1,每次输入模型的序列包含1 024个位点,网络的预测结果包含在经过Softmax函数之后的预测向量中,取概率最高的位置所对应的类别作为预测类型。
3.3 评价指标
本文使用平均填补准确率()与每个批次序列填补准确率的标准差()对模型结果进行评估。缺失位点的位置可以通过记录缺失基因型矩阵位点确定,通过比较原始基因型数据与填补结果数据在缺失位置上是否保持一致来计算吻合率,所得到的结果称为填补准确率,表示为:

3.4 有效性实验
为了验证FCSA的填补性能与泛化能力,将它应用到三款开源动物的参考面板中,针对不同物种进行训练后,分别在10%、20%、30%的缺失率下,在不同物种的测试集上进行验证,计算所有批次序列的填补准确率的平均值,每个物种在不同缺失率下的与如表1所示。由表1可以看出,对于这三组数据集,FCSA对Dog数据集的拟合效果最好,在三种缺失率下的分别为93.69%、93.52%、93.41%。实验结果表明,随着缺失率的升高,FCSA依然具备较好的健壮性,依次仅下降0.17和0.11个百分点。但是Chicken与Pig两个数据集上的却低于Dog,而在整体较低的Chicken数据集中,FCSA在10%的缺失率下的依然到达了84.62%。不同物种基因平均填补准确率上的差异说明了物种间基因数据的复杂程度不同,也验证了设计多组物种数据进行对比实验的必要性。
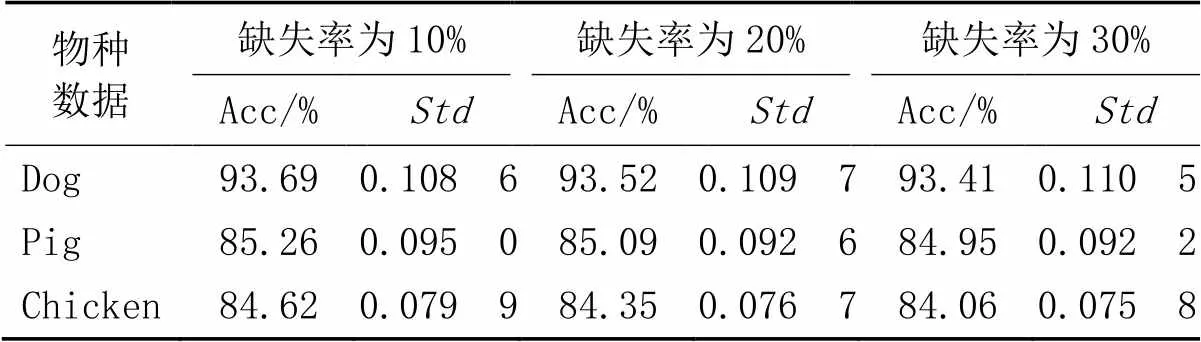
表1 三个物种数据在不同缺失率下的平均填补准确率
3.5 模型对比实验
3.4节的实验结果显示,FCSA在Dog数据集上表现较好,但在Chicken数据集上表现较差。为了验证FCSA在基因型填补中的先进性,分别在Dog与Chicken数据集上设计对比实验,并选取SCDA[12]、AGIC[13]、U‑net[15]作为基线,实验结果如表2所示。表2中Dog数据集的填补结果表明,FCSA在不同缺失率下的达到了四种模型中的最高值;SCDA与AGIC的相当,但是FCSA在每个批量序列填补准确率的分散程度略高于SCDA方法,而AGIC虽然较高,但是批次序列的填补准确率比较分散,较高;除此之外,U‑net的与表现最差。由Chicken数据集的结果可以看出,不同方法对该数据集的整体低于Dog数据集,这表明不同物种位点间具有不同的相关模式,分散及异质程度不同。本文FCSA方法在不同缺失率下的仍达到了四种模型中的最高值,批次序列填补准确率的分散程度也最低,这表明在复杂的连锁关系中,两种融合模块能够较充分地提取并融合局部与全序列信息,这是其余三种单模式填补方法所不具备的。
综上可知,本文FCSA方法在平均填补准确率达到了最高水平,且批量序列填补准确率的分散程度较低;SCDA的平均填补准确率低于本文方法;AGIC的平均填补准确率仅次于本文方法,但是批量序列的填补准确率在两组数据上都较为分散,导致部分序列填补准确率极高而部分序列填补准确率极低,这是模型建模序列长度太短所导致的;U-net方法在两组数据集上均表现出了最差的效果,这说明将图像重建的思想应用于基因型填补领域并不是最合适的。

表2 Dog和Chicken数据集上的对比实验结果
3.6 消融实验
消融实验主要是探究两种融合模块的有效性。针对填补问题,引入了如下的对比实验:FCSA-a表示两融合模块不采用注意力机制;FCSA-b表示编码器融合模块中先捕捉局部信息再整体建模并融合;FCSA-c表示解码器融合模块中先整体建模再重建局部信息并融合。
针对消融实验的网络模型,在连锁关系不易捕捉的Chicken数据集上进行实验,为了验证FCSA在较大缺失率下的健壮性,仅测试缺失率为30%的情况,平均填补准确率与批量序列的填补准确率如表3、图5所示。可以看出,仅仅在基于卷积的网络模型中堆叠注意力机制对提高整个填补准确率作用不大,甚至效果会更差,这也反映了FCSA模型中编码器融合模块与解码器融合模块设计的合理性。这也说明本文模型设计的融合模块有助于捕获基因位点之间的关联关系,能够有效地对缺失位点进行填补。

表3 消融实验结果
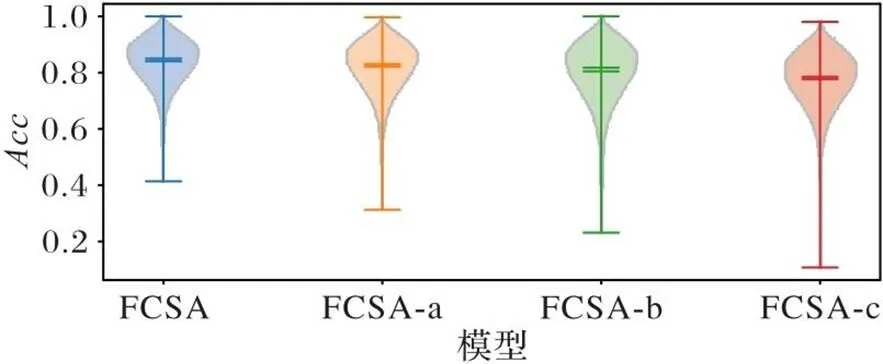
图5 批量序列的平均填补准确率分布
4 结语
现有的基于深度学习的基因型填补方法不能有效建模并计算长序列位点间的连锁关系,造成了总体填补准确率低、批量序列填补准确率较分散的问题。因此,本文设计了一种融合卷积与自注意力机制的填补方法FCSA,改进原始Transformer模块并提出两种融合模块来构建网络模型,两种融合模块能兼顾局部与全局位点的关联模式,在不丢失位置信息的前提下对缺失序列进行特征提取与重建。通过对比实验验证了FCSA在相关模式不同的物种序列中均提高了填补准确率、降低了批量序列填补准确率的分散度,并通过消融实验验证了两种融合模块设计的有效性。进行多物种对比实验时,意识到可以实现基于多物种数据的预训练模型,但因硬件限制并没有展开设计,今后可以在多物种基因数据的预训练及模型蒸馏上开展进一步研究。
[1] International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome[J]. Nature, 2001, 409(6822): 860-921.
[2] LI Y, WILLER C, SANNA S, et al. Genotype imputation[J]. Annual Review of Genomics and Human Genetics, 2009, 10: 387-406.
[3] WIGGINTON J E, CUTLER D J, ABECASIS G R. A note on exact tests of Hardy-Weinberg equilibrium[J]. AJHG: The American Journal of Human Genetics, 2005, 76(5): 887-893.
[4] PEI Y F, LI J, ZHANG L, et al. Analyses and comparison of accuracy of different genotype imputation methods[J]. PLoS ONE, 2008, 3(10): No.e3551.
[5] ZHANG Z, DRUET T. Marker imputation with low-density marker panels in Dutch Holstein cattle[J]. Journal of Dairy Science, 2010, 93(11): 5487-5494.
[6] 李乐义,邵东东,丁向东,等.SNP芯片基因型填充至测序数据的策略[J].中国科技论文,2016,11(12):1431-1436. (LI L Y, SHAO D D, DING X D, et al. Research on genotype imputation from SNP chip data to whole-genome sequence data[J]. China Sciencepaper, 2016, 11(12): 1431-1436.)
[7] MARCHINI J, HOWIE B, MYERS S, et al. A new multipoint method for genome-wide association studies by imputation of genotypes[J]. Nature Genetics, 2007, 39(7): 906-913.
[8] HOWIE B N, DONNELLY P, MARCHINI J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies[J]. PLoS Genetics, 2009, 5(6): No.e1000529.
[9] LI Y, WILLER C J, DING J, et al. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes[J]. Genetic Epidemiology, 2010, 34(8): 816-834.
[10] SCHEET P, STEPHENS M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase[J]. AJHG: The American Journal of Human Genetics, 2006, 78(4): 629-644.
[11] BROWNING B L, BROWNING S R. Genotype imputation with millions of reference samples[J]. AJHG: The American Journal of Human Genetics, 2016, 98(1): 116-126.
[12] CHEN J, SHI X. Sparse convolutional denoising autoencoders for genotype imputation[J]. Genes, 2019, 10(9): No.652.
[13] ISLAM T, KIM C H, IWATA H, et al. A deep learning method to impute missing values and compress genome-wide polymorphism data in rice[C]// Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies — Volume 3. Setúbal: SciTePress, 2021: 101-109.
[14] 曹一珉,蔡磊,高敬阳.基于生成对抗网络的基因数据生成方法[J].计算机应用,2022,42(3):783-790. (CAO Y M, CAI L, GAO J Y. Gene data generation method based on generative adversarial network[J]. Journal of Computer Applications, 2022, 42(3): 783-790.)
[15] 殷力. 基于深度学习的基因型填充方法研究[D]. 北京:中国科学院大学, 2020: 30-40.(YIN L. Genotype imputation method based on deep learning[D]. Beijing: University of Chinese Academy of Sciences, 2020: 30-40.)
[16] VASWANI A, SHAZEER N, PARMAR N, et al. Attention is all you need[C]// Proceedings of the 31st International Conference on Neural Information Processing Systems. Red Hook, NY: Curran Associates Inc., 2017: 6000-6010.
[17] BA J L, KIROS J R, HINTON G E. Layer normalization[EB/OL]. [2023-02-23].https://arxiv.org/pdf/1607.06450.pdf.
[18] YANG W, YANG Y, ZHAO C, et al. Animal-ImputeDB: a comprehensive database with multiple animal reference panels for genotype imputation[J]. Nucleic Acids Research, 2020, 48(D1): D659-D667.
Genotype imputation algorithm fusing convolution and self-attention mechanism
CHEN Jionghuan1,2, BAO Shengli1,2*, WANG Xiaofei1,2, LI Ruofan1,2
(1,,610213,;2,100049,)
Genotype imputation can compensate for the missing due to technical limitations by estimating the sample regions that are not covered in gene sequencing data with imputation, but the existing deep learning-based imputation methods cannot effectively capture the linkage among complete sequence loci, resulting in low overall imputation accuracy and high dispersion of batch sequence imputation accuracy. Therefore, FCSA (Fusing Convolution and Self-Attention), an imputation method that fuses convolution and self-attention mechanism, was proposed to address the above problems, and two fusion modules were used to form encoder and decoder to construct network model. In the encoder fusion module, a self-attention layer was used to obtain the correlation among complete sequence loci, and the local features were extracted through the convolutional layer after fusing the correlation to global loci. In the decoder fusion module, the local features of the encoded low-dimensional vector were reconstructed by convolution, and the complete sequence was modeled and fused by self-attention layer. The genetic data of multiple species of animals were used for model training, and the comparison and validation were carried out on Dog, Pig and Chicken datasets. The results show that compared to SCDA (Sparse Convolutional Denoising Autoencoders), AGIC (Autoencoder Genome Imputation and Compression) and U-net, FCSA achieves the highest average imputation accuracy at 10%, 20% and 30% missing rate. Ablation experimental results also show that the design of the two fusion modules is effective in improving the accuracy of genotype imputation.
genotype imputation; convolution; self-attention; fusion module; full sequence modeling
1001-9081(2023)11-3534-06
10.11772/j.issn.1001-9081.2022111756
2022⁃11⁃24;
2023⁃02⁃06;
中国科学院“西部青年学者”项目(RRJZ2021003)。
陈炯环(1998—),男,山东潍坊人,硕士研究生 ,主要研究方向:机器学习、大数据系统、大规模数据分析; 鲍胜利(1973—),男,安徽黄山人,研究员,博士,主要研究方向:软件工程、大数据智能; 王啸飞(1997—),男,湖南慈利人,硕士研究生,主要研究方向:机器学习、推荐算法; 李若凡(1997—),男,甘肃兰州人,硕士研究生,主要研究方向:机器学习、时序预测。
TP391.1
A
2023⁃02⁃09。
This work is partially supported by “Western Young Scholars” Project of Chinese Academy of Sciences (RRJZ2021003).
CHEN Jionghuan, born in 1998, M. S. candidate. His research interests include machine learning, big data system, large-scale data analysis.
BAO Shengli, born in 1973, Ph. D., research fellow. His research interests include software engineering, big data intelligence.
WANG Xiaofei, born in 1997, M. S. candidate. His research interests include machine learning, recommendation algorithm.
LI Ruofan, born in 1997, M. S. candidate. His research interests include machine learning, time series forecasting.
