Ni2 B(001)析氧反应性能的第一性原理
2023-11-25虞江涛高洪涛
虞江涛,高洪涛
(青岛科技大学 化学与分子工程学院,山东 青岛 266042)
电解水是产生清洁能源最有效的方法之一。在现有的电解水技术中,由阴极析氢反应(HER)和阳极析氧反应(OER)组成的水电解技术是提供清洁,高纯度且可持续的产氢途径的高效手段[1-2]。因此,开发具有高活性、高稳定性、高导电性的OER 电催化剂是发展燃料电池和金属-空气电池的一个关键步骤[3-4]。到目前为止,Ir基和Ru基金属化合物是目前最先进的OER 电催化剂[5-6]。然而,这些贵金属的稀缺性和高成本严重阻碍了它们在能量转换和存储设备中的大规模应用。因此,迫切需要开发一些耐用、高效和低成本非贵金属OER 电催化剂。
作为能量转换和存储材料,过渡金属硼化物,例如硼化镍(Ni2B)引起了越来越多研究者的兴趣[7-8]。由于硼原子的半径较小,在形成化合物时很容易填入到金属晶格中的空隙,所以Ni2B 中的金属原子可以在很大程度上保持原有的金属键连接模式。此外,B的电子数、电负性、电离能、独特的外层电子排布形式,以及其与过渡金属原子之间电子转移数量的多样性决定了过渡金属硼化物中化学键的成键方式和成键强度,并最终增强了Ni2B 的OER 催化性能。
本研究基于DFT 的第一性原理对Ni2B(001)晶面的OER 催化性能进行探究,探讨了Ni2B(001)晶面的稳定性以及其OER 反应活性。
1 计算方法
1.1 模型构建
体相Ni2B(I4/mcm)理论结构模型见图1。
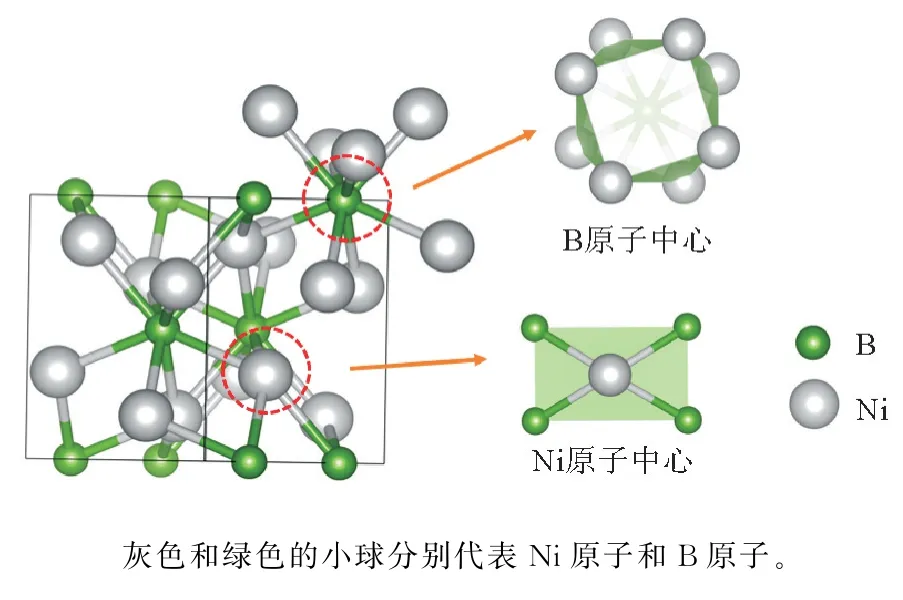
图1 Ni2 B的体相结构图Fig.1 Diagram of the bulk phase structure of Ni2 B
由图1可见,8个镍原子分布在B原子周围,形成1个以B原子为中心8个Ni原子为顶点的球体。4个B原子分布在Ni原子周围,形成1个以Ni原子为中心的五面体。这种交错排列的结构有利于电子在镍原子和硼原子之间互相转移,从而增强其表面OER 活性。Ni2B中的Ni Ni键是具有良好导电性的金属键,能促进电化学反应的进行。
1.2 计算参数
所有的计算都是基于密度泛函理论(DFT)且采用 Vienna Ab initio Simulation Package(VASP)[9-10]实现的。电子与离子的相互作用是用投影增强波(PAW)[11-12]电位来描述的,广义梯度近似(GGA)以Perdew-Burke-Ernzerhof(PBE)函数的形式来描述交换相关能量[13]。同时,Ni2B(001)表面和吸附原子之间的范德瓦尔斯相互作用采用DFT-D3分散校正方案进行校正,并考虑了自旋极化的影响。平面波基础的截断能量为400 eV,所有的几何结构都被完全弛豫和优化,直到对原子的作用力小于0.02 eV·Å-1,电子自洽性的收敛标准被设定为10-5eV。在几何优化过程中,布里渊区的K 点取值3×3×1,超胞单元为2×2×1。为了避免相邻晶胞之间的相互作用,在Z方向上采用了大于15Å 的真空度。
Ni2B表面结构的稳定性通常用表面能的相对大小来表示。表面能越小,结构越稳定,表面能计算如公式(1)所示[14]。
其中Es,relax和Es,unrelax是弛豫和未弛豫的表面的总能量,Eb是体相晶胞每个Ni2B 分子的平均能量,eV,N是体相中单位分子的数量,A是所考虑的表面面积。宏观上,表面能通常以J·m-2为单位表示。在DFT 计算中,表面能以eV·Å-2为单位。
2 结果与讨论
2.1 Ni2 B晶面活性
OER 反应发生在催化剂的表面,因此有必要研究表面的特性,其中表面的活性取决于其原子结构。低米勒指数的表面比高米勒指数的表面更稳定[15],因此,低米勒指数的表面是首选。在这项工作中研究的表面是沿低指数平面(001)、(010)、(100)、(110)、(011)、(101)和(111)来标记定义的。研究发现具有3层厚度的Ni2B模型足以模拟Ni2B表面属性。真空度实验表明,在Z方向表面添加20Å 的真空度足以减少相邻表面模型间的相互作用[16]。表1显示了Ni2B 7个晶面的洁净表面能。从表1可以看出,(001)面的能量是最低的,也是最稳定的。因此,选择最稳定的Ni2B(001)晶面进行OER 模拟研究。

表1 Ni2 B的不同晶面的洁净表面能Table 1 Clean surface energy of different crystal surfaces of Ni2 B
2.2 析氧反应机理
对表面机理的理论研究能更好的探明Ni2B(001)晶面的OER 催化活性。正如NØRSKOV等[17]所提出的,OER 是一个缓慢的四电子转移过程,给定催化剂的OER 活性受四电子转移步骤的吉布斯自由能控制。根据NØRSKOV 等[18]提出的理论模型,OER 在碱性条件下反应的机理如下面所示[19]:
式中*为催化剂的表面活性位点,带*的分子代表吸附状态。
其中吉布斯自由能变(ΔG)由公式(2)计算,
ΔEDFT代表通过DFT 计算得到的总能量,EZP代表通过振动频率计算得到的自由分子的零点能量,TS代表熵。吸附分子的零点能量和熵的变化,可以从DFT 振动频率计算得出。在4步反应的吉布斯自由能变中,吉布斯自由能变最大值定义为ΔGOER,可以表示为
在标准条件下,当U与标准氢电极(SHE)等于0时,理论上的过电势ηOER 与ΔGOER的关系:
理想地,当ηOER=0时,ΔGOER为1.23 eV,在理想状态下,OER催化剂的吉布斯自由能变为1.23 eV[20-21]。
2.3 析氧反应活性
本工作研究了掺杂前后的OER 性能变化,见图2。如图2所示,水电解产生的OH-首先吸附在表面吸附活性位点形成吸附态的OH*,随后OH*被去质子化形成O*,接下来生成的O*与另一个OH-结合形成OOH*中间体。在最后一步反应中,OOH*中间体经过去质子化产生O2从表面逸出,并在表面生成新的活性位点。
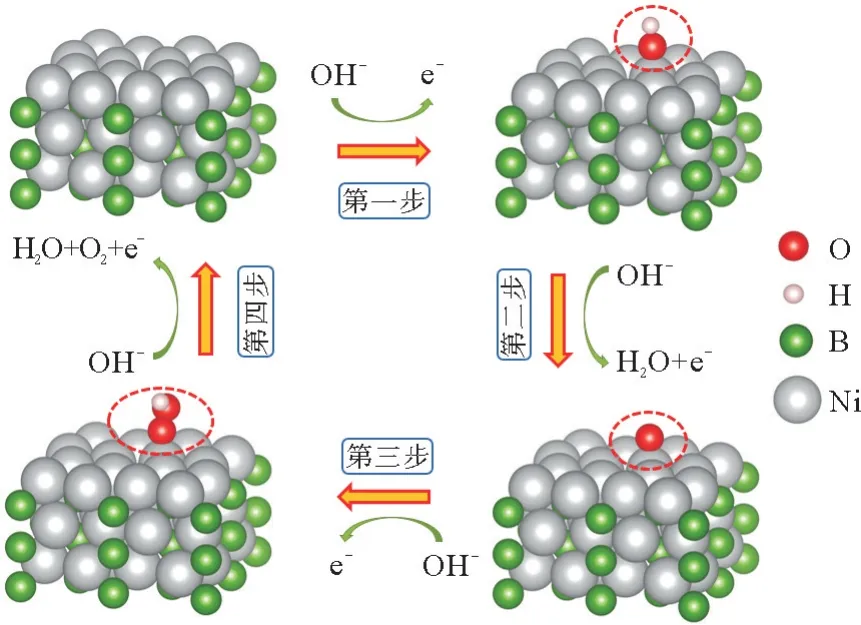
图2 Ni2 B(001)表面OER反应四电子机理示意图Fig.2 Schematic diagram of the four-electron mechanism of the OER reaction on the Ni2 B(001)surface
由反应机理可知,OER 共有4 个基本反应步骤。利用OER 过程中每个基本步骤的吉布斯自由能变(ΔG)变化来体现OER 活性强弱,其中具有最大ΔG值的步骤代表电势决定步骤(potential determining step,PDS)[17]见图3。如图3所示,步骤3为整个OER 反应的决速步骤,从中间体*O 至*OOH 的反应步骤代表整个OER 过程的PDS。在整个反应过程中,U=0 V 时,自由能曲线呈阶梯式上升状态。在理论过电势η=1.23 V 时,第一步、第二步和最后一步反应都是自发的,而决速步骤依然有明显的能量势垒,需要更高的过电势去克服。理论计算表明,决速步骤理论最小过电势η=1.500 V。由此可知当U增加到2.730 V(对应于1.500 V 的理论超电势)时,OER 决速步骤的ΔG值降低至0 eV,表明整个OER 过程可以在该电势之上自发进行。

图3 Ni2 B(001)表面OER反应四电子步骤的ΔG 图Fig.3 ΔG of the four-electron step of the OER reaction on the Ni2 B(001)surface.OER on the Ni2 B(001)surface
掺杂是提高材料物理和化学性能的最广泛的方法之一,它可以通过重新分配电荷、形成悬挂键、改变配位数等方法有效改变ΔG。本工作采用单原子掺杂的方法,对Ni2B(001)晶面单个Ni原子进行替代掺杂,从而达到降低OER 反应过电势,提高其催化活性的目的。过渡金属M 掺杂前后Ni2B(001)晶面OER 4个步骤的吉布斯自由能见表2。由表2可知,与原Ni2B(001)相比,在掺入过渡金属M 后,反应决速步骤的吉布斯自由能变(ΔG3)减小,从而使催化活性得到提高。其中Ni2B-Cr(001)的吉布斯自由能最低,所需的过电势最低(η=0.857 V),催化活性也最高。故而在以上掺杂过程中,过渡金属原子Cr掺杂效果最好。
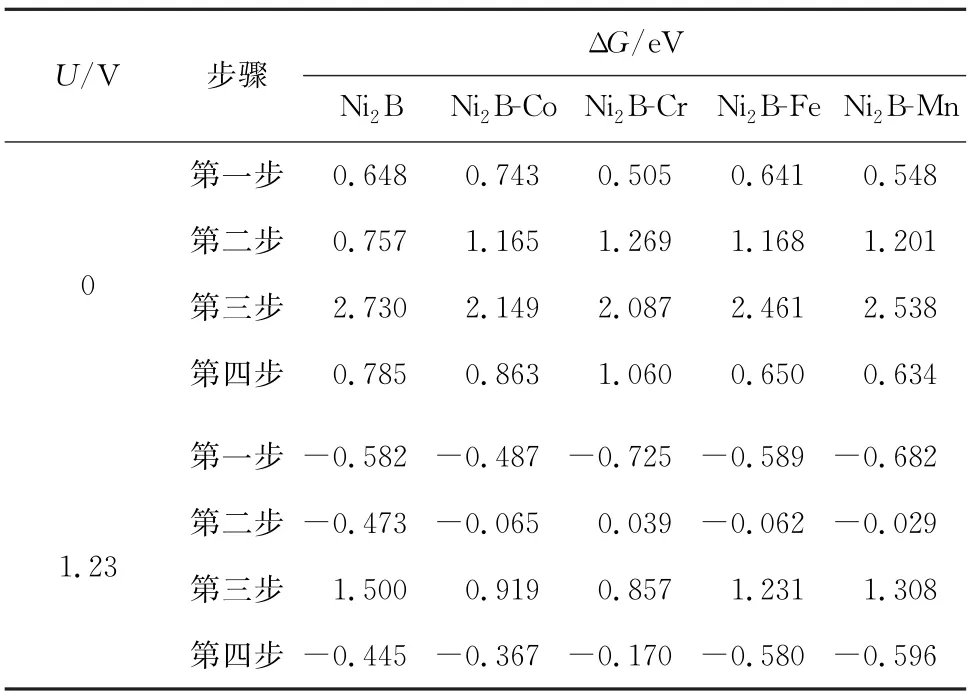
表2 过渡金属M掺杂前后Ni2 B(001)晶面OER 4个步骤的吉布斯自由能Table 2 Gibbs free energy(eV)of the four steps of the Ni2 B(001)facet OER before and after doping with transition metal M
2.4 电荷分析
为了更深入的探究过渡金属M 的掺杂能提高Ni2B(001)OER 性能的原因,对掺杂前后的电荷转移进行了研究。掺杂前后bader电荷分布见表3。
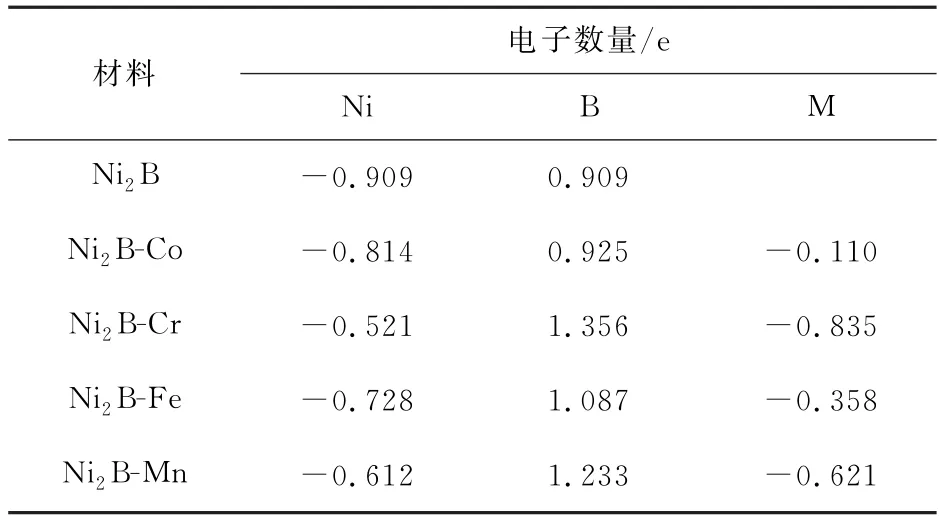
表3 掺杂前后bader电荷分布Table 3 Charge distribution of bader before and after doping
由表3 bader电荷分析可知,未掺杂时,表面B原子周围电荷是由Ni原子转移得到的。掺杂后,由于引入了新的过渡金属原子M,过渡金属M 的电子也开始向B 原子转移,转移的数量依次为:0.110,0.835,0.358和0.621 e。其中相较于掺杂前,B原子得到的电荷依次为:0.016,0.447,0.178,0.324 e。由此可见,掺杂后表面的M 原子周围电荷向内部的B原子转移。因此,M 原子周围则产生了更多的电子空穴,M 原子附近的电子空穴更有利于富电子的OER 中间体的吸附,从而使得表面的OER 活性增强。Cr掺杂后对表面电荷分布的影响最大,原子周围的电子空穴最多,OER 反应中间体在表面的吸附也更稳定,更有利于OER 反应的发生。这与掺杂前后的吉布斯自由能研究结果一致,从而佐证了该结论是可靠的。
2.5 态密度和d带中心分析
一般来说,材料的导电性和催化活性都与催化剂的电子结构特性密切相关。根据玻尔兹曼方程,材料的导电性与它在费米能级附近的载流子密度成正比。费米能级附近的载流子密度越高,材料的电导率就越高[22]。因此,对掺杂前后Ni2B 的电子结构特进行研究,以探索其电催化活性。Ni2B(001)过渡金属M 掺杂前后表面的dos图见图4。
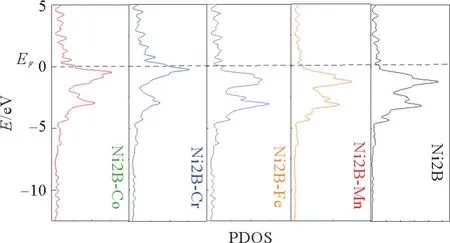
图4 Ni2 B(001)过渡金属M 掺杂前后表面的dos图Fig.4 Dos diagram of the surface before and after Ni2 B(001)transition metal M doping
从图4可以看出,在费米能级(EF)附近有多个能带相互重叠,表明Ni2B 具有良好的导电性。在掺入M 后,表面的镍原子与掺杂原子的3d轨道明显重叠,在费米能级附近有一些轻微的突出峰,这表明M 的掺入可以有效激活M 原子附近的镍原子的催化活性,从而增强Ni2B 电催化剂的导电性和OER 性能。
在分析OER 反应的过程中考虑d带中心的影响是非常有用的。d带的中心[23]可以用态密度来计算:
其中x是能量,p(x)是态密度。根据d带中心模型,从一个表面到另一个表面的吸附能量的变化与d带中心相对于费米能量的上移相关。更多的上移表明有可能形成更多的空反键合状态,从而促进形成更强的结合能。因此,OER 催化性能的强弱可以用d带中心的上移来描述。通常,占据费米能级的电子越多,催化剂的活性越高。因此,吸附位点的d带中心越靠近费米能级,活性位点和表面吸附态原子之间的结合强度就越强,OER 反应就越容易在表面发生[24-25]。过渡金属M 掺杂后的表面原子的d带中心Ni2B-Co(001)(-1.30 eV)、Ni2B-Cr(001)(-1.24 eV)、Ni2B-Fe(001)(-1.40 eV)和Ni2BMn(001)(-1.28 eV),明显高于未掺杂的Ni2B(001)(-1.46 eV)的表面原子,且更靠近费米能级。根据d带中心理论,过渡金属M 掺杂后的表面表现出更优异的电化学催化性能。而且相较于其他3种掺杂情况,Ni2B-Cr(001)的d带中心更接近费米能级,从而有着更优异的OER 性能,这也和吉布斯自由能研究结果一致(图5)。以上理论研究结果表明,过渡金属原子M 的掺杂能够提高Ni2B 的电导率和析氧性能。
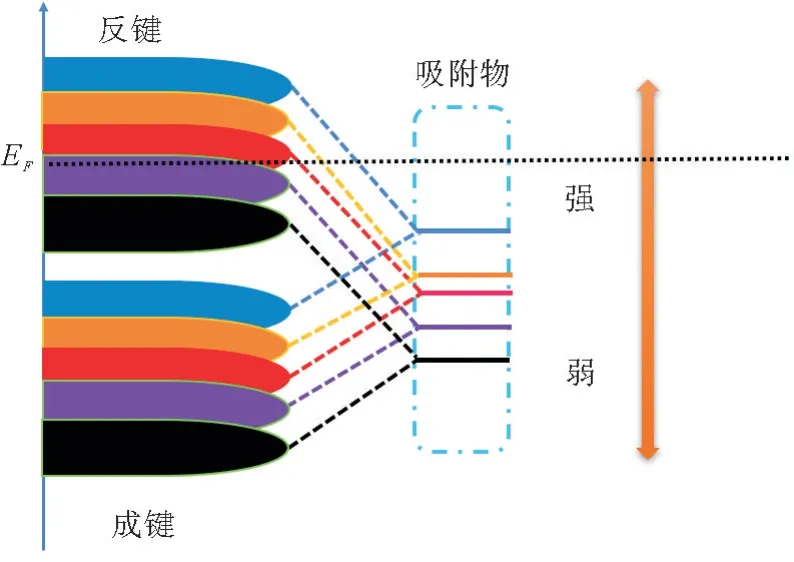
图5 d带中心位置以及催化剂表面和吸附物之间相互作用的相应示意图Fig.5 Position of the centre of the d-band and the corresponding schematic diagram of the interaction between the catalyst surface and the adsorbate
3 结论
通过基于第一性原理的密度泛函理论计算,本研究从结构稳定性,过渡金属原子掺杂前后表面电荷变化,OER 的催化活性和态密度d带中心变化几个方面研究了Ni2B(001)晶面的OER 催化性能。研究发现,Ni2B(001)有着较好的OER 性能。为了进一步提升Ni2B(001)的OER 催化活性,选择了四种具有相同周期的过渡金属(Co、Cr、Fe和Mn)进行表面单原子掺杂取代。过渡金属M 掺杂后表面的电子特性的研究也表明,掺杂后表面B原子获得了更多的电荷,费米能级上出现了新的峰值,d带中心向费米能级移动,Ni2B(001)表面的OER 性能得到了很大提升。研究表明,Ni2B是一种潜在的非贵金属OER 催化剂。
