Co-MoP/MoS2/Cu的制备及其电催化析氢性能
2023-11-25纪晓阳邓晓燕
纪晓阳,邓晓燕
(青岛科技大学 环境与安全工程学院,山东 青岛 266042)
自工业革命以来,人类社会的发展就一直依赖于煤和石油等传统化石燃料,然而随着科技的进步和各种新技术、新产业的发展,人类对化石能源的消耗越来越大,能源与环境问题日益加剧。科学家一直在寻找一种新型的能源利用方式,以解决能源和环境危机,实现可持续发展。氢气(H2)是一种可以实现脱碳的新型能源,具有能量密度高(~120 MJ·kg-1)[1]且零碳排放的优点,在“2030 碳达峰、2060碳中和”的大背景下备受关注。
目前国际范围内普遍将氢气按照生产来源及生产过程中的排放情况分为灰氢、蓝氢及绿氢。灰氢是指通过化石燃料燃烧产生的氢气,生产途中会排放二氧化碳,目前是全球氢气产量最主要的来源;蓝氢是指天然气通过蒸汽甲烷重整或自热蒸汽重整制得的氢气,由于生产过程中使用了碳捕捉、储存等技术,温室气体被捕获,可以实现低碳排放;绿氢是指利用可再生能源,如风能、太阳能等通过电解水制氢,实现零碳排放。基于以上3种氢气的特点,电解水制绿氢被认为是能够实现碳中和的终极能源利用方式。然而想要实现高效电解水制绿氢,需要高性能的电催化剂来促进析氢反应(HER)。目前,最有效的电催化剂是铂、钯、铱或钌等贵金属基催化剂,但贵金属高昂的价格阻碍了它们的工业化应用[2],寻找廉价、高性能、长寿命的贵金属替代材料非常重要,科学家们在这个热门研究领域已经进行了大量探索[3-6]。过渡金属磷化物(TMPs)由于其独特的物理化学性质,包括高导电性、地球储量丰富[7],成为一种很有前途的贵金属催化剂替代品[8]。其中,磷化钼基材料展现出了优异的析氢性能,引起研究者们的广泛兴趣[9-13]。
本研究以简便的水热合成法和气相磷化法合成了高性能的电催化析氢电极,并通过Co元素的加入改善了电极表面形貌,提高了电极材料的电催化析氢性能。电化学测试表明,在电流密度为10 mA·cm-2时,过电位为60 mV,具有良好的电催化析氢活性。在大电流密度下具有良好的稳定性,在300 mA·cm-2的电流密度下连续工作24 h后,性能无明显下降,能更好地满足工业化的要求。
1 实验部分
1.1 试剂与仪器
泡沫铜,苏州科盛和金属材料;钼酸铵、硫脲、六水合氯化钴、次亚磷酸钠、无水乙醇,盐酸,国药集团化学试剂有限公司。
电化学工作站,CHI760E 型,上海辰华仪器有限公司;控温管式炉,JYG-C01型,上海精宏实验设备有限公司;电热鼓风干燥箱,DHG-9030A 型,上海一恒科学仪器有限公司;数控超声波清洗器,KQ500GDB型,昆山市超声仪器有限公司;X 射线衍射仪,Rigaku D/MAX/2500PC 型,日本理学公司;场发射扫描电子显微镜,HITACHI SU8010型,日立科学仪器公司;X 射线光电子能谱(XPS),Shimadzu AXIS SUPRA 型,日本岛津公司,利用Al Kα辐射作为X 射线源。
1.2 实验方法
1.2.1 泡沫铜的前处理
购买来的泡沫铜表面可能会存在氧化层或加工时残存的有机物,需要进行前处理后使用。将泡沫铜裁剪成1.5 cm×4 cm 的长方形,放置于盛有无水乙醇的烧杯中,超声处理10 min,用去离子水冲洗,随后放入盛3 mol·L-1稀盐酸的烧杯中再次超声清洗10 min,放入烘箱,50℃烘干备用。
1.2.2 MoS2/Cu及Co-MoS2/Cu前驱体的合成
称取0.05 mmol钼酸铵,0.1 mmol硫脲溶解于30 mL去离子水中,混合均匀后移入40 mL聚四氟乙烯内衬的反应釜中,将处理过后的泡沫铜浸入,密封后移入烘箱,140℃下反应12 h,得到MoS2前驱体[14]。Co-MOS2前驱体的合成方法与MoS2前驱体相同,仅在其基础上另称取0.05 mmol六水合氯化钴溶解于40 mL去离子水中。
1.2.3 MoP/MoS2/Cu及Co-MoP/MoS2/Cu的合成
以次亚磷酸钠为磷源,采用气相磷化的方法对前驱体进行磷化。分别将3 mmol次亚磷酸钠及前驱体放入管式炉的上游及下游,在氩气气氛下400℃高温烧结2 h,利用次亚磷酸钠受热分解产生的磷化氢气体对前驱体进行磷化。得到MoP/MoS2/Cu电极及Co-MoP/MoS2/Cu电极。
1.3 Co-MoP/MoS2/Cu系列电极的电化学测试
所有的电化学测量都是在标准三电极体系中,以1 mol·L-1的KOH 溶液为电解液,固定温度为25℃,使用CHI760E 电化学工作站进行的。将实验所得到的电极裁剪成特定的形状,使用铂片电极夹固定,用作工作电极;汞/氧化汞(Hg/Hg O)电极用作参比电极。在所有的实验中,对电极都采用碳棒电极,所有绘图所使用的电位都转换为可逆氢电极电位,转化公式:
其中:ERHE为相对于可逆氢电极的电位;E为相对于参比电极的电位;EHg/HgO为汞氧化汞参比电极相对于可逆氢电极的电位;溶液pH 值在本研究中为固定值14。
1.3.1 电化学极化曲线的测量
为保证电化学测试期间电极相对稳定,在测试极化曲线之前先对电极进行30次循环伏安活化,以保证电极达到相对稳定的状态。设定窗口电压为-0.9~-1.4 V,扫描速率5 mV·s-1,重复测试3次以上,以确保避免偶然误差。
1.3.2 电化学交流阻抗谱(EIS)的测量
固定过电位为300 mV,即设定测试电压为-1.222 7 V,设定电压频率高频1 000 Hz,低频0.01 Hz,得到各电极在过电位300 mV下的交流阻抗谱。
1.3.3 电化学活性面积(ECSA)的评估
电化学活性面积通过循环伏安法测量双电层电容间接评估,测试须在非法拉第区间进行,设定电压窗口为-0.45~-0.55 V,分别测量扫描速率为5,10,15,20 mV 的循环伏安曲线,对电流密度的差值和扫描速率进行线性拟合,所得直线斜率的二分之一即为双电层电容,电化学活性面积与双电层电容呈正比例相关。
1.3.4 稳定性测试
稳定性测试采用计时电流法进行评估,固定测试电压为-1.5 V,测试时间24 h,测试电流随时间的变化,得到时间-电流曲线。
2 结果与讨论
2.1 Co-MoP/MoS2/Cu系列电极的表征及分析
图1是前驱体及电极的SEM 照片。如图1(a)所示,MoS2/Cu前驱体由大量纳米球堆积而成,并且在纳米球表面密布着片状的突起;但是经过高温气相磷化后,多个相邻的纳米球在高温作用下烧结为粒径较大的纳米球,并且原本存在于表面的片状结构消失,MoP/MoS2/Cu电极表面变为凹凸不平的类似于荔枝皮的结构(如图1(b)所示)。磷化后的电极与未经磷化的前驱体相比,表面形貌在高温气相磷化过程中被破坏,会阻碍析氢性能的进一步提升。为了使磷化后的电极具有更大的比表面积,尝试对在合成过程中引入Co元素。如图1(c)所示,引入Co元素后的Co-MoS2/Cu 前驱体呈不规则球状,杂乱无章地堆积在一起,且表面分布少量刺状突起,经过高温气相磷化后,Co-MoP/MoS2/Cu电极表面烧结为粒径稍大的表面密布着刺状突起的圆球(如图1(d)所示),Co的加入使得原本较为平滑的表面出现密集的刺状突起,提高了材料表面的粗糙程度,有利于活性位点的暴露,能有效提升其析氢性能。通过BET 比表面积测试可以得到材料的N2吸附等温线(图2)和比表面积数据(表1),Co加入后,材料的比表面积从0.772 3 m2·g-1增加到了0.901 6 m2·g-1,更粗糙的表面形貌给材料带来了更大的比表面积,增大了材料与电解液的接触面积,有利于催化性能的提高。
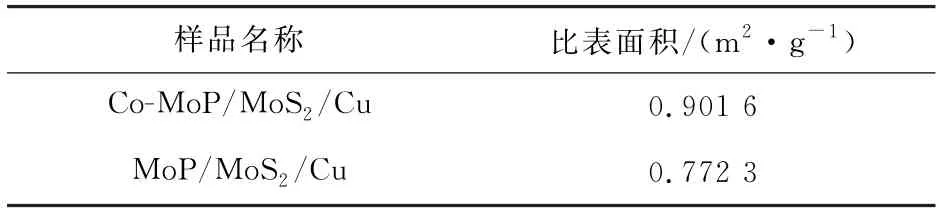
表1 根据BET结果计算所得材料的表面积Table 1 Surface area of the resultant materials calculated from BET results

图1 前驱体及Co-MoP/MoS2/Cu系列电极的SEM 照片Fig.1 SEM images of precursor and Co-MoP/MoS2/Cu series electrodes
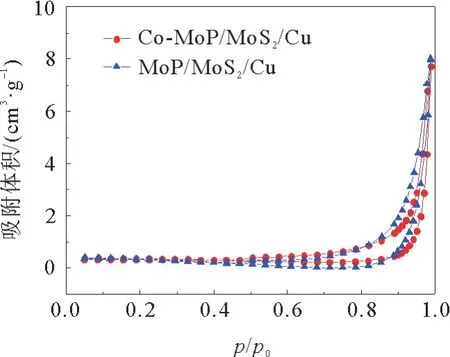
图2 N2吸附等温线Fig.2 N2 sorption isotherms of different samples
图3是Co-MoP/MoS2/Cu电极不同放大倍数下的SEM 照片。如图3(a)所示,低放大倍数下,可以清楚的看到Co-MoP/MoS2均匀地生长于泡沫铜骨架之上,且粒径均匀分布在10μm 左右。在高放大倍数下,可以清晰地看到纳米球表面均匀分布的刺状突起。该种结构比表面积较大,也与BET 结果相符,能够使电解液充分与电极接触,从而使更多的活性位点发挥作用,提升电极的析氢性能。

图3 Co-MoP/MoS2/Cu电极的SEM 照片Fig.3 SEM images of Co-MoP/MoS2/Cu electrodes
元素映射图像和EDS能谱如图4和图5所示。合成过程中所涉及的Mo、S、P 元素均匀分布在纳米球表面。Cu元素仅集中在泡沫铜基底上,如图4(c)右下角部分区域所示。Co元素在元素映射图像中仅显示出极少量的亮点,结合图5的EDS能谱,其中并没有像Cu和Mo元素一样明显的峰,各元素含量如表2 所示,Co的含量仅为0.406%,以上表征方式共同说明Co元素并没有参与构成催化剂。Co的存在仅影响了MoS2前驱体的生长,形成了不同于图1(a)形貌的前驱体,如图1(c)所示,不同形貌的前驱体在后续的高温气相磷化中生成了不同形貌的磷化物。

表2 催化剂中各元素含量Table 2 Content of each element in thecatalyst

图4 Co-MoP/MoS2/Cu的EDS元素映射图像Fig.4 EDS elemental mapping images of Co-MoP/MoS2/Cu

图5 Co-MoP/MoS2/Cu的EDS能谱Fig.5 EDS spectrum of Co-MoP/MoS2/Cu
图6是二硫化钼前驱体和MoP/MoS2/Cu电极及Co-MoP/MoS2/Cu 电极的XRD 谱图。可以清楚的看到在所有谱图中的43.3°,50.5°及74.1°处均出现3个较明显的峰,这是泡沫铜基体所造成的衍射峰,与Cu 的标准卡片(PDF#65-9026)相对应。MoP/MoS2/Cu 电极及Co-MoP/MoS2/Cu 电极的谱图中,在45.1°和46.1°处出现两个较为明显的衍射峰,与Cu3P的标准卡片(PDF#65-3628)相对应,这是泡沫铜基体在气相磷化时与磷化氢气体接触,导致金属铜被磷化所导致的。除此之外并无其他明显衍射峰的存在,但是在MoP/MoS2/Cu 电极和Co-MoP/MoS2/Cu电极XRD 谱图的25°左右出现一个不明显的无定形峰,透射电镜照片及电子衍射结果(图7)表明材料无明显晶格条纹,电子衍射图也没有清晰的衍射环的出现,而是一个弥散状的衍射环,据此推测生长在泡沫铜基底上的MoS2和MoP均为非晶态。在水热合成过程中,由于Co元素的加入,影响了MoS2的生长,导致材料呈现无定形态,这也有可能导致材料的电催化析氢性能提升[15]。
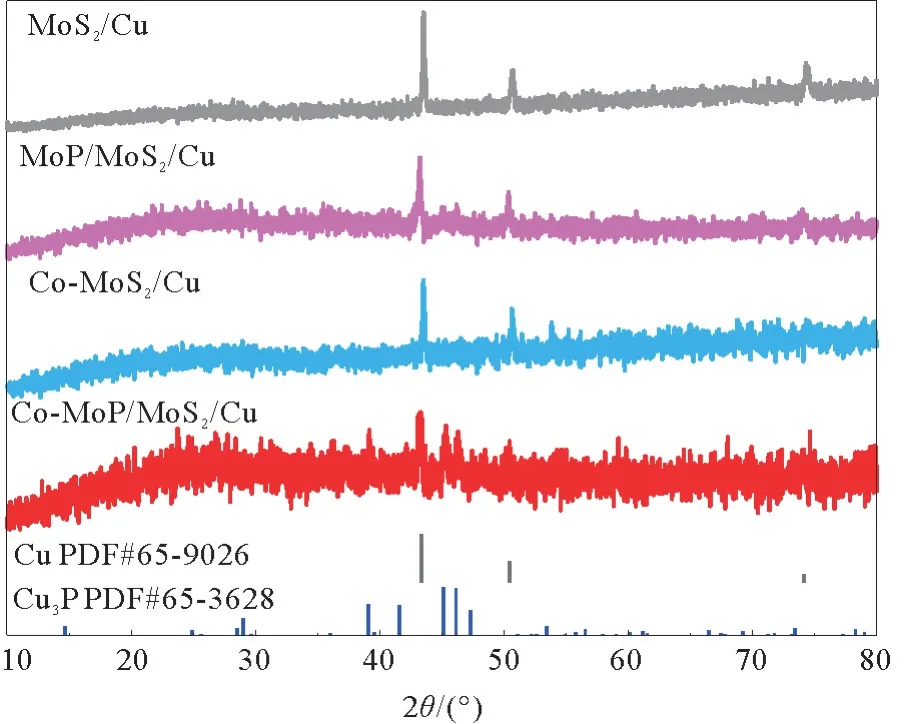
图6 前驱体及Co-MoP/MoS2/Cu系列电极的XRD谱图Fig.6 XRD patterns of precursor and Co-MoP/MoS2/Cu series electrodes

图7 HRTEM 和SAED照片Fig.7 HRTEM and SAED images
为了进一步确认电极材料中的元素组成和化学态,对电极进行了XPS 测试,图8 是Co-MoP/MoS2/Cu电极的XPS谱图。图8(a)是XPS全谱,证明了电极材料含有Cu、Mo、S、P元素,Cu元素来自于泡沫铜基底,O元素来自于合成过程中电极材料与空气接触时发生的氧化反应,值得注意的是,XPS全谱中并没有出现明显的Co元素的峰。为了进一步精确探究材料表面各元素的状态,对材料进行了高分辨XPS测试,如图8(b)~(e)。图8(b)是Co 2p的高分辨谱图,所得结果与全谱类似,并没有出现明显的Co元素峰,综合EDS及元素映射的结果,推测在水热合成过程中Co元素可能并不直接参与构成催化剂,但是在SEM 图像中可以清晰的看出合成过程中Co 元素的加入对材料形貌的影响,可以推测,合成过程中加入的Co元素引入并不能直接参与材料的合成,而是会在水热合成过程中间接影响材料在泡沫铜基底上的生长过程,导致材料形貌上的变化,使得比表面积变大,从而导致材料的电化学析氢性能提升。

图8 Co-MoP/MoS2/Cu电极的XPS谱图Fig.8 XPS spectrum images of Co-MoP/MoS2/Cu electrodes
Mo 3d XPS 谱图如图8(c)所 示,在229.5 和230.9 eV 出现两个弱峰,对应着Moδ+(0<δ<4),可能是气相磷化过程中,还原性气体PH3使得MoS2前驱体中的Mo(Ⅳ)部分还原成较低价态的Mo和P 的化合物。而位于232.8和233.9 eV 的两个特征峰则表明Mo元素在电极材料中主要以Mo(Ⅳ)氧化态的形式存在,可能是MoS2前驱体未全部磷化,部分Mo元素仍以MoS2的形式存在的原因。除此之外还有两个位于235.8和236.5 eV相对最弱的峰,分别对应着Mo O3(可能是电极材料暴露于空气中形成)或(可能是合成材料所用的钼酸盐的残留)的Mo(Ⅵ)态的Mo 3d结合能[16]。
如图8(d)所示,S 2p谱图中的163.1和161.9 eV 处的峰,分别对应于S 2p1/2和S 2p3/2结合能[17]。P 2p 谱图中,如图8(e)所示,130.3 和129.4 eV 的峰对应于P 2p1/2和P 2p3/2结合能,134.3 eV 的峰值对应着P O,可能是材料暴露在空气中被氧气氧化导致的[18]。
图8(f)是Cu的2p谱图,在932.2和952.0 eV附近的峰归因于泡沫铜被气相磷化后形成的Cu3P和泡沫铜基底的共同作用,另外在942.9和962.2 eV 附近出现2个卫星峰。934.5和954.8 eV 附近的峰是由于材料暴露在空气中形成的CuO所造成的。
2.2 电化学测试及分析
采用稳态线性扫描伏安法(LSV),在25℃1.0 mol·L-1KOH 电解液中,以5 mV·S-1的扫描速率测试得到各电极的极化曲线,研究材料的电催化析氢性能。图9(a)黑色曲线是未经任何处理的泡沫铜的极化曲线,其在1.0 mol·L-1KOH 电解液中并未表现出明显的析氢活性,而代表Co-MoP/MoS2/Cu电极的红色曲线表现出了最优的析氢性能,在10 mA·cm-2的电流密度下过电位为60 mV,在450 mV 过电位下达到的电流密度可高达700 mA·cm-2,大电流下表现出了优异的析氢性能,已经接近商业化的铂碳催化剂(绿色曲线所示),更加贴近工业电解水的要求。通过极化曲线得到Tafel斜率,如图9(b)所示,对电极材料的析氢动力学进行研究,MoP/MoS2/Cu 电极和Co-MoP/MoS2/Cu 电极的Tafel 斜率几乎一致,在120 mV·dec-1左右,只有MoS2/Cu前驱体(228 mV·dec-1)和Co-MoS2/Cu 前驱体(240 mV·dec-1)的二分之一左右,说明经过气相磷化后的电极材料在析氢反应动力学上更快速。EIS阻抗谱图用来研究电极材料电子转移的动力学,见图10。如图10所示,在-300 mV 的测试电位下,Co-MoP/MoS2/Cu电极的EIS阻抗谱显示出最低的电荷转移电阻(Rct),表明Co-MoP/MoS2/Cu电极的界面电阻最小,电子转移过程更快[19]。

图9 前驱体及Co-MoP/MoS2/Cu系列电极的HER极化曲线及Tafel斜率Fig.9 HER polarization curve and Tafel plot of precursor and Co-MoP/MoS2/Cu series electrodes

图10 前驱体及Co-MoP/MoS2/Cu系列电极的EIS阻抗谱图Fig.10 EIS diagram of precursor and Co-MoP/MoS2/Cu series electrodes
前驱体及Co-MoP/MoS2/Cu系列电极的循环伏安曲线见图11。如图11(a)~(d)所示,利用电流密度中间值,与循环伏安扫描测试中的扫描速率进行线性拟合,获得的斜率的一半对应于双电层电容Cdl值[20]。电流密度中间值与扫描速率的线性拟合结果如图12所示,结果表明Co-MoP/MoS2/Cu电极的Cdl值最高,为319 mF·cm-2,其电化学活性面积也最大。结合上文的SEM 照片BET 测试等,可以推测Co的加入导致电极材料比表面积增大,进而使电化学活性面积增大,最终使电极材料电化学析氢性能获得提升。

图11 前驱体及Co-MoP/MoS2/Cu系列电极的循环伏安曲线Fig.11 CV curves of precursor and Co-MoP/MoS2/C series electrodes

图12 前驱体及Co-MoP/MoS2/Cu系列电极的双电层电容Fig.12 Electric double layer capacitance of precursor and Co-MoP/MoS2/Cu series electrodes
催化剂的稳定性对于能量存储和转换技术至关重要。使用计时电流法评估Co-MoP/MoS2/Cu电极的耐久性24 h,见图13。如图13所示,在此期间析氢电流密度稳定,没有观察到明显可识别的性能衰减,这表明电催化剂具有优异的稳定性,有应用于工业化的电解水制氢的潜质。
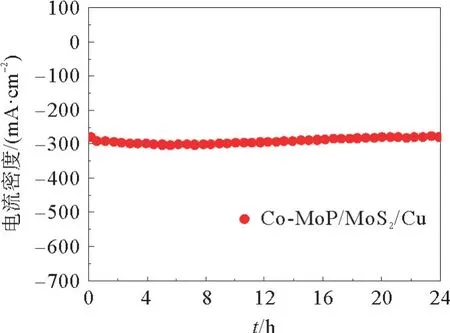
图13 Co-MoP/MoS2/Cu电极的计时电流曲线Fig.13 Timing current curve of Co-MoP/MoS2/Cu electrodes
3 结语
通过简单可控的水热合成法及气相磷化法,在泡沫铜上,以二硫化钼为前驱体,合成了自支撑的电催化析氢电极,并通过Co的加入调控电极材料表面形貌,使材料形貌由荔枝状变为表面密布刺状突起的球状,获得了更大的比表面积和电化学活性面积。该电极在10 mA·cm-2的电流密度下过电位为60 mV,在450 mV 过电位下电流密度可高达700 mA·cm-2,大电流下表现出了优异的析氢性能,且稳定性良好,可长时间工作,更加贴近工业电解水的要求。
