乙酸乙酯与ε-CL-20不同晶面的微观作用机制
2022-11-04谢五喜廖奕玫陈伶媛段晓惠
游 婷,谢五喜,廖奕玫,陈伶媛,段晓惠
(1.西南科技大学 环境友好能源材料国家重点实验室,四川 绵阳 621010;2.西安近代化学研究所,陕西 西安 710065)
引 言
六硝基六氮杂异伍兹烷(CL-20)自1987年问世以来,就因其高能量密度特性,在炸药、固体推进剂、发射药等领域引起广泛关注。在晶体特征方面主要包括高品质CL-20结晶和降感[1-3]、CL-20的多晶型及其晶变机理和抑制方法[4-8]等。这些研究主要通过溶液结晶过程来实现,因此结晶溶剂的选择至关重要。
目前大部分研究主要关注溶剂对ε-CL-20晶体形貌和晶型的影响,如采用分子动力学(MD)模拟技术,通过校正的附着能(AE)模型,预测了ε-CL-20在单一溶剂或混合溶剂中的晶体形貌[9-13]。主要采用溶剂层与晶面层的相互作用能来对真空中的附着能进行校正,从而预测溶液中可能的形貌。另外,动力学因素逐步引入晶体形貌的预测中,螺旋生长模型和2D成核模型等机械生长模型的发展极大提高了理论预测的可靠性。Lovette和Doherty结合螺旋生长和2D成核模型,提出了Lovette-Doherty (L-D) 模型,该模型可以预测全部过饱和度区间内的晶面生长[14]。Shim等[15]采用L-D模型准确预测了不同溶解度和多种溶剂中ε-CL-20的晶体形貌。ε-CL-20是当前溶剂诱导转晶研究最多的炸药,研究表明[16-18],溶剂、温度、过饱和度、添加剂、晶种、搅拌方式、溶剂/非溶剂的种类和用量均会影响CL-20不同晶型的析出。溶剂对晶体形貌和晶型的影响,均涉及溶剂与不同晶面的相互作用,但目前大多数研究主要体现的是溶剂的整体效应,而对原子分子层次上的具体作用机制关注很少。因此,本研究以ε-CL-20为研究对象,选用乙酸乙酯(EA)作溶剂。采用MD模拟技术对EA溶剂与ε-CL-20不同晶面的微观作用机制进行研究,所得结果可为溶液结晶中炸药晶体形貌和晶型控制提供理论依据,亦可加深对溶液结晶过程和机理的理解。
1 模型构建与模拟方法
1.1 晶面特性
从剑桥有机晶体数据库(CCDC)库中导出ε-CL-20的晶体结构(CCDC编号为PUBMUU17),构建其晶胞模型。利用Materials Studio材料模拟平台中的Forcite模块,对其进行分子力学(MM)优化。设置如下优化参数:Compass力场,力场指定电荷,“Fine”精度,“Smart”方法,“Ewald”方法计算静电相互作用和范德华力,优化晶胞参数。分子模拟精度对力场依赖度高,Compass力场已在RDX[19]、HMX[20]、CL-20[11,12]等硝胺类炸药的分子和晶体模拟中得到成功应用。
真空中的晶体形貌预测采用AE模型,由Morphology模块来完成,力场和计算精度与结构优化相同,“Ewald”法计算静电相互作用,“Atom based”法计算范德华力。基于形貌预测结果,对主要生长面进行切割,建立晶面模型。为了表征晶面的几何拓扑结构和粗糙度(比如扭折和台阶结构),计算了不同晶面的Connolly面积。采用Materials Studio软件“Tool”工具中的“Atom Volume & Surface”,计算精度Ultrafine,格点间隔0.15Å,Connolly半径1.000Å。为了更准确地评判晶面的粗糙度,定义了K参数:
K=Sconn/Sunit
(1)
式中:Sconn为晶面单元的Connolly面;Sunit为晶面单元的表面积。
不同晶面的带隙计算采用DMol3方法。首先将ε-CL-20晶胞按晶面进行切割,切割层厚度为2层,为避免额外的自由边界效应,在晶面上加30Å的真空层。对切割得到的晶面模型进行结构优化,GGA/PBE泛函,精度为“Fine”,DFT-D校正采用TS-Grimme方法,随后对能带结构进行分析,即可得到带隙值。
1.2 溶剂与晶面的相互作用
采用双层界面模型研究溶剂与晶面的相互作用,模型构建分3步(见图1):(1)参照EA密度(0.90g/cm3)构建包含400个分子的溶剂层,并对其进行MM结构优化;(2)对晶面进行切割并扩展成超晶面(扩展倍数见图2),获得相应的晶面层模型并进行MM结构优化;(3)由溶剂层和晶面层构建双层界面模型,并在溶剂层上方添加50Å的真空层。
模型构建好之后,在固定晶面层的情况下进行后续计算。首先对双层界面模型进行MM结构优化。上述所有MM结构优化的参数设置均与单胞相同。为了消除界面应力,先进行退火分子动力学(MD)模拟,参数设置如下:能量参数(Compass力场、“Fine”精度和“Ewald”计算静电和范德华力)和动力学参数(退火循环数为10;初始温度为300.0K;中间循环温度为500.0K;每个循环的加热梯度为5;每个梯度的模拟步数为5000;总步数为500000;每个循环均进行结构优化)。对退火后的界面模型再进行常规MD模拟:NVT系综,控温方法Anderson,积分步长1.0fs,总模拟时间为500ps,Ewald方法计算范德华和静电相互作用,每隔20000步输出一帧。基于最终的平衡结构采用式(2)计算溶剂层和晶面的结合能Es:
Es=-(Etot-Esur-Esol)/n
(2)
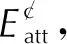
(3)
(4)
结合能的计算体现了溶剂层与晶面作用的整体效应,为了更清晰地“看见”其作用细节,本研究模拟了单个溶剂分子与晶面的吸附作用。在Adsorption Locator模块中基于Metropolis方法搜索低能吸附位点,主要参数为“Simulated annealing”、“Fine”精度、Compass力场、静电和范德华力的计算方法分别为“Ewald”和“Atom based”。
为了进一步研究不同晶面的吸附量和吸附热,采用Sorption模块对其吸附作用进行模拟研究。首先将ε-CL-20晶胞扩展为2×2×2的超胞,然后分别按6个晶面进行切割,切割厚度为1层。为避免额外的自由边界效应,构建了双晶面层模型,两层晶面间的真空层设为10Å。模拟方法仍为Metropolis,固定压力为101.325kPa,分别采用“Ewald & Group”和“Atom based”方法计算静电相互作用和范德华力,基于巨正则系综(μVT,即系统的化学势μ、体积V和温度T保持恒定)计算吸附在晶面上的溶剂分子数和吸附热。以上所有计算均在Materials Studio 4.0中完成[21]。
2 结果与讨论
2.1 晶面特性
优化后的晶胞参数及其对应的实验值和相对误差列于表1。从表1可以看出,采用Compass力场的分子力学结构优化,其最大误差为b轴的2.59%。模拟误差均小于5%,在分子模拟方法可接受的范围内。因此,在后续的计算中均采用Compass力场。
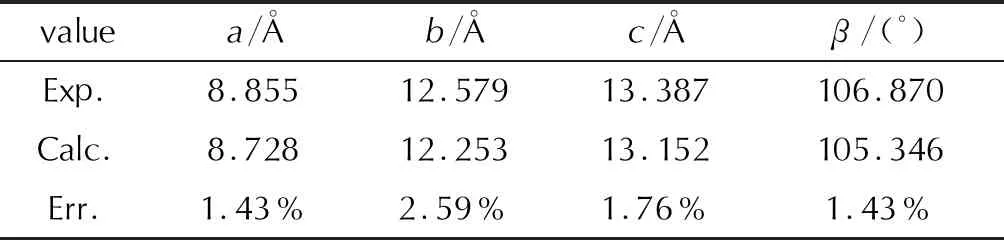
表1 ε-CL-20模拟前后的晶胞参数及相对误差Table 1 Cell parameters and relative errors before and after ε-CL-20 simulation
AE模型预测ε-CL-20的真空形貌类似梭状,由6个独立晶面组成,其特征参数列于表2中。 晶面大小顺序为:(0 0 2)<(0 1 1)<(1 0 -1)<(1 1 0)<(1 1 -1)<(1 0 1)。最重要的晶面为(0 1 1)面,其显露面占总显露面的39.59%,其次是(1 1 0)面(26.04%),(1 0 1)面的占比最小,仅2.80%。各晶面显露面的大小顺序为:(0 1 1)<(1 1 0)<(1 0 -1)<(0 0 2)<(1 1 -1)<(1 0 1)。显露面的大小受晶面的生长速率控制,生长速率越快,显露面越小,反之亦然,而生长速率则与附着能Eatt的绝对值成正比。
从表2可以看出,显露面最大的(0 1 1)面,其附着能绝对值最小(363.01kJ/mol),生长速率最慢,是形态学上最重要的晶面。显露面最小的(1 0 1)面,具有最大的附着能绝对值(470.21kJ/mol)。附着能由静电能和范德华力组成,各个晶面的静电能均占据主导作用,范德华力的贡献较小。从表2附着能的计算结果可以预测不同晶面生长速率的相对顺序,即:(0 1 1)<(1 0 -1)<(1 1 0)<(0 0 2)<(1 1 -1)<(1 0 1)。和显露面的大小顺序相比,除(1 0 -1)和(1 1 0)两个晶面的顺序相反之外,其他完全相同。而造成该差异的原因为不同晶面显露面的大小不仅与附着能有关,还与晶面的多重度有关,这两个因素共同决定显露面的大小。

表2 ε-CL-20不同晶面特征参数Table 2 Parameters of different crystal faces of ε-CL-20
不同晶面的带隙计算结果为:(0 0 2)>(1 0 1)>(0 1 1)>(1 0 -1)>(1 1 -1)>(1 1 0)。可以看出,对表面最为平坦的(0 0 2)面,其带隙最高,比该晶面带隙稍低的是(1 0 1)。带隙最低的晶面是(1 1 0),其次为(1 1 -1),文献[22, 23]将带隙值与撞击感度进行关联,即带隙越大,撞击感度越低。根据该关系,(0 0 2)和(1 0 1)为撞击感度较低的晶面,而(1 1 0)和(1 1 -1)为撞击感度较高的晶面。但目前对根据带隙值预测撞击感度尚存在争议[24, 25]。
图2形象地显示了ε-CL-20晶面的表面结构。可以看出,6个晶面无论在拓扑结构还是显露的化学基团方面均有显著差异。6个晶面上显露的化学基团主要是硝基,即最外层均为硝基氧原子,伴随着次外层亚甲基上的氢原子,但这些基团在不同晶面上的密度和排布方式差异很大。比如(1 1 -1)晶面上的硝基显著突出晶面,呈一行一行的排列方式,行与行之间留下了深且宽的空隙。(1 0 1)晶面呈波浪状排列,形成了仅次于(1 1 -1)面的空隙结构。(0 0 2)晶面上仅硝基氧原子分布在晶面上,且氧原子的密度较小,晶面较平坦。基团在晶面的分布差异也带来了拓扑结构的不同,比如台阶和扭折的大小和多少,而这些结构也是质点进入晶格促使晶体生长的位置。从表2计算的K值可以看出,6个晶面粗糙度的顺序为:(1 0 -1)>(1 1 -1)>(0 1 1)>(1 0 1)>(1 1 0)>(0 0 2)。即粗糙度最大为(1 0 -1)面,其次为(1 1 -1)面,(0 0 2)面的粗糙度最小,这与图2的表面结构相吻合。

图2 ε-CL-20不同晶面的表面结构(晶面长宽高的扩大倍数)Fig.2 Interfacial structures of different faces of ε-CL-20 (magnification of length, width and height)
2.2 EA与晶面的相互作用
2.2.1 EA层与晶面的相互作用


表3 EA溶剂与ε-CL-20不同晶面的结合能(单位:kJ/mol)Table 3 Binding energies of EA solvent with different faces of ε-CL-20 (unit: kJ/mol)
为进一步考察溶剂与不同晶面的作用特性,图3显示了界面上沿晶面法向方向(z轴)EA的相对浓度(c)。可以看出,EA与所有晶面均构成了一定厚度的溶剂界面层。对沿z轴方向厚度为26.15Å的(1 1 -1)面,在约22Å处就可观察到溶剂分子,随后其相对浓度随距离(d)缓慢上升,在d约30Å时达到最大(约3.6),随后衰减到平均浓度(溶剂的体浓度)。可以看出,EA分子已进入到(1 1 -1)晶面内约4Å的深度,构成一个厚度约10Å的溶剂界面层,有较多溶剂分子与晶面发生相互作用。对沿z轴方向厚度为19.45Å 的(0 0 2)晶面,没有溶剂分子进入晶面内部,在晶面上约20Å处才能观察到极少量的EA分子。随着d的增加,EA相对浓度快速上升到3.2,随后快速衰减到平均浓度,溶剂界面层厚度约为4Å。与(1 1 -1)面相比,与(0 0 2)面发生相互作用的溶剂分子少,结合紧密度低。这与两个晶面的拓扑结构(图2)和结合能的计算结果(表3)一致。(1 1 0)和(0 0 2)面一样,晶面较为平坦,没有溶剂分子进入晶面内部,其他3个晶面均有少量溶剂分子进入晶面。但所有晶面的溶剂界面层厚度、界面层中溶剂的相对浓度均小于(1 1 -1)面,这也印证了溶剂与(1 1 -1)面的结合能最大。

图3 界面上沿ε-CL-20不同晶面法向方向(z轴)EA分子的相对浓度(c)分布(图中所标数值为各晶面在z轴方向上的厚度)Fig.3 Relative concentrations of EA molecules along the normal direction (z axis) of different faces of ε-CL-20 (depth of each habit face along z axis)
2.2.2 单个EA分子在不同晶面上的吸附作用
相互作用能的计算体现了溶剂层与晶面的整体效应,为了更清晰地“看见”其作用细节,本研究模拟了单个溶剂分子与晶面的吸附作用。吸附能的计算结果列于表4。由表4可以看出,为了更好地吸附在晶面上,EA分子发生了一定的变形,所需能量定义为“变形能”。EA在不同晶面上的变形能较为接近,绝对值最大的为(0 1 1)和(1 1 0)面的7.91kJ/mol,最小的为(1 0 1)面的6.20kJ/mol。从表4的模拟结果还可发现不同晶面吸附能绝对值的大小顺序为:(1 1 -1)>(1 0 -1)>(1 1 0)>(1 0 1)>(0 1 1)>(0 0 2)。该顺序与溶剂层和晶面的结合能顺序除(1 0 -1)和(1 1 0)发生交换外,其他完全相同,即结合能和吸附能绝对值最大的均为(1 1 -1),而最小的均为(0 0 2)。造成(1 0 -1)和(1 1 0)顺序差异的主要原因应为表面积的大小,(1 0 -1)面的表面积(168.19Å2)小于(1 1 0)面(195.52Å2)。因为吸附能仅仅考虑一个溶剂分子与晶面活性位点的作用情况,而表3中的结合能计算的是溶剂层与晶面作用的整体效应。二者相互印证,更为详细地说明了溶剂和不同晶面的作用差异。此外,从吸附能的高低也可初步判断晶面的相对生长速率。一般来讲,吸附能越高,晶面的生长速率越快。从表4可以预测,生长最快的晶面为(0 0 2),生长最慢的(1 1 -1),其次是(1 0 -1)。其他3个晶面的生长速率较为接近。基于吸附能预估的晶面相对生长速率,与校正后的附着能预测结果基本一致。
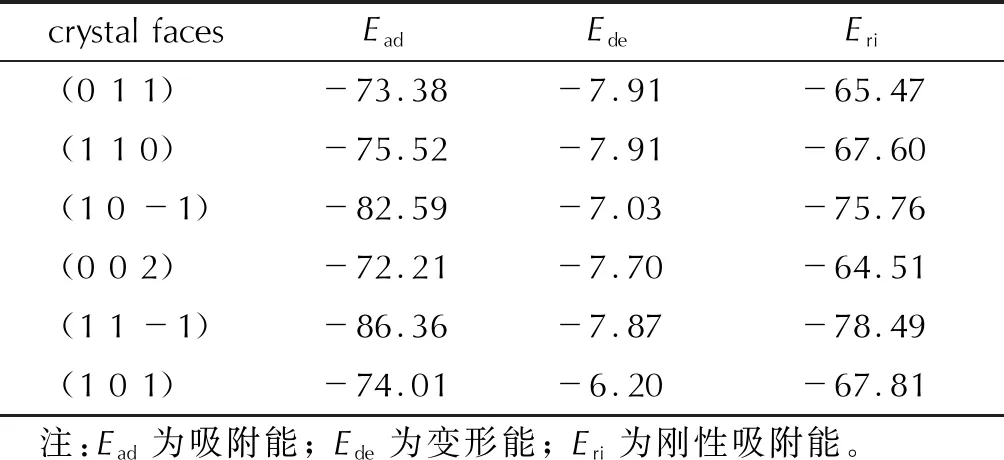
表4 EA分子在ε-CL-20不同晶面上的吸附能(单位:kJ/mol)Table 4 Absorption energies of EA molecule on different faces of ε-CL-20(unit:kJ/mol)
EA在各个晶面的吸附图见图4和图5。为了清晰起见,图中仅标出了部分键长小于3Å的氢键。
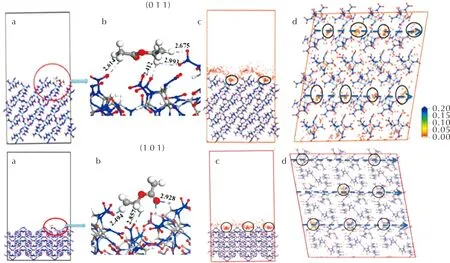
图4 EA分子在(0 1 1)和(1 0 1)面上的吸附作用[a:沿x轴方向吸附界面结构的侧视图;b:EA分子在界面上的具体作用图像;c:沿x轴方向吸附位点密度场的侧视图;d:沿z轴方向吸附位点密度场的俯视图。b中蓝色虚线表示氢键、黑色圆圈为吸附概率较大的吸附位点;d中蓝色虚线显示晶面上的吸附通道)Fig.4 Adsorption of EA molecule on (0 1 1) and (1 0 1) faces (a: side view of interfacial structure along x axis;b: interaction of EA with ε-CL-20 on crystal faces; c: side view of density field of adsorption sites along x axis;d: top view of density field of adsorption sites along z axis. Hydrogen bonds denote as dotted lines in green, adsorption sites with high probability as black cycle, and adsorption channel as dotted lines in blue)
图4显示了EA与(0 1 1)和(1 0 1)面的作用情况。EA分子与这两个晶面均显示了较好的亲和力,其甲基和亚甲基氢与裸露于晶面的硝基氧原子形成了多个氢键,见图4(b),键长范围在2.4~3.0Å,以及羰基氧原子与硝基氧原子形成的O…O相互作用等。两个晶面的吸附能分别为-73.38和-74.01kJ/mol。图4(c)和(d)为吸附位点的密度场,色标从红到蓝表示吸附几率依次增加。可以看出吸附概率较大的绿色区域、吸附概率极低的红色区域以及吸附概率较低的黄色区域在晶面上的分布情况。几个吸附概率较大的区域位于晶面上的台阶或扭折处,见图4(c)和(d)中的黑色圆圈,这些台阶或扭折结构在晶面周期排列形成沿x轴的吸附通道,见图4(d)中的蓝色虚线,有利于吸附分子的附着。此外亦可看出,EA分子已通过表面的台阶进入晶面内部,形成结合紧密的界面结构。
图5显示了EA在另外4个晶面上的吸附情况。在这4个晶面上,EA分子与晶面上的ε-CL-20分子,仍主要通过C—H…O弱氢键、O…O等相互作用结合在一起。其中,(1 0 -1)是表面粗糙度最大的一个晶面,晶面上有众多的台阶结构,形成了蓝色虚线所示的沿y轴的吸附通道,非常有利于EA分子在晶面上的吸附。从吸附位点图可看出,EA分子在该晶面上的吸附位点较多;(1 1 -1)面的粗糙度仅次于(1 0 -1)。和(1 0 -1)面的台阶结构相比,该晶面有更大的袋状空隙。这些袋状空隙在晶面上周期排列,形成了较大体积的通道,可以很好地容纳吸附分子,使吸附分子和周围的晶面发生更紧密的接触。从表4吸附能的计算结果也可看出,EA分子在该晶面的吸附作用最强,表明该晶面受溶剂的影响最大,这和前面溶剂和晶面结合能计算结果一致。
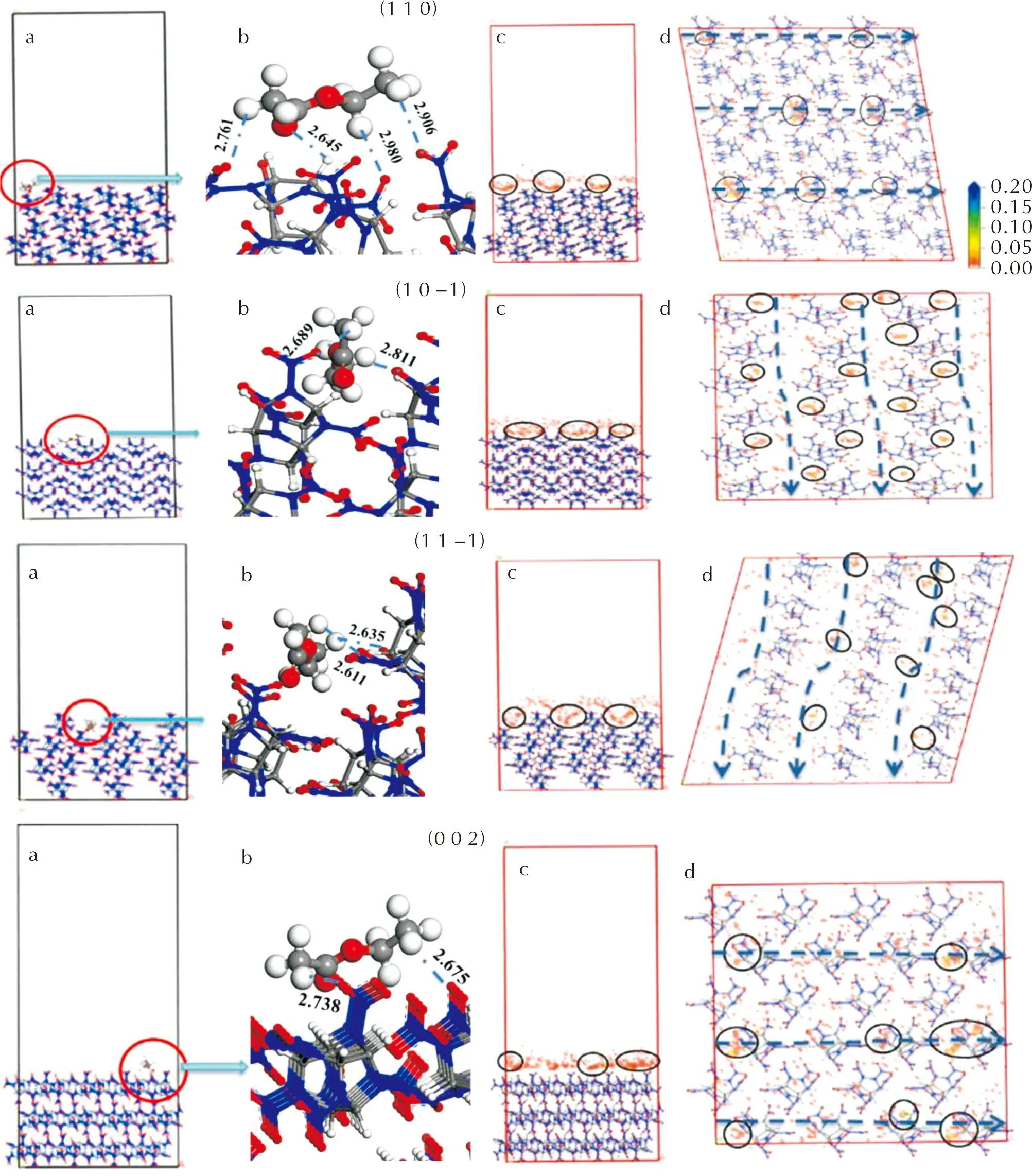
图5 EA分子在4个不同晶面上的吸附作用(a:沿y轴方向吸附界面结构的侧视图;b:EA分子在界面上的具体作用图像;c:沿y轴方向吸附位点密度场的侧视图;d:沿z轴方向吸附位点密度场的俯视图)Fig.5 Adsorption of EA molecule on four faces (a: side view of interfacial structure along y axis; b: interaction of EA with ε-CL-20 on four faces; c: side view of density field of adsorption sites along y axis; d: top view of density field of adsorption sites along z axis).
作为6个晶面中表面最为平坦的(0 0 2)晶面上,高几率吸附位点形成沿x轴的吸附通道。吸附分子EA与晶面的相互作用发生在晶面之上,造成二者的结合强度降低,这与EA分子在该晶面上的吸附能绝对值最低相一致(见表4)。(1 1 0)晶面的吸附情况介于(1 0 -1)、(1 1 -1)和(0 0 2) 之间,EA分子主要吸附在晶面上较浅的扭折处,未进入晶面内部。这是因为(1 1 0)面较为平坦,其粗糙度仅大于最为平坦的(0 0 2)面(见图2和表2),吸附分子与晶面的相互作用也主要发生在晶面之上。
2.2.3 EA分子在晶面上的吸附量和吸附热
采用Sorption模块模拟得到的饱和吸附结构和吸附分子数及等量吸附热见图6和表5。可以看出,(1 1 -1)吸附的EA分子数最多(28个),其次是(1 0 1)(24个),而(0 0 2)吸附的EA分子数最少,仅5个,其他3个晶面吸附的EA分子数相当。吸附分子数的多少主要由晶面拓扑结构(图2)和晶面大小(表2)决定。(1 1 -1)和(1 0 1)的表面积分别为201.35 和215.71Å2,远大于(0 0 2)的106.95Å2,同时表面分布有大的袋状空隙 [(1 1 -1)] 或“V”形台阶结构[(1 0 1)],相对于(0 0 2)平坦的表面结构,能更好地容纳EA分子。此外,所有晶面的吸附热均为正值,结合表4中负的吸附能值,说明EA在这些晶面上的吸附均为放热过程。从表5还可看出,吸附EA分子数最多的(1 1 -1)晶面,其等量吸附热最小,而吸附EA分子数最小的(0 0 2)面等量吸附热最大,即吸附分子个数与等量吸附热呈负相关。
图6 ε-CL-20晶体6个晶面对EA分子的饱和吸附结构(吸附的EA分子以绿色显示)Fig.6 Saturation adsorption of six faces of ε-CL-20 for EA molecules (EA molecules denoted as green)
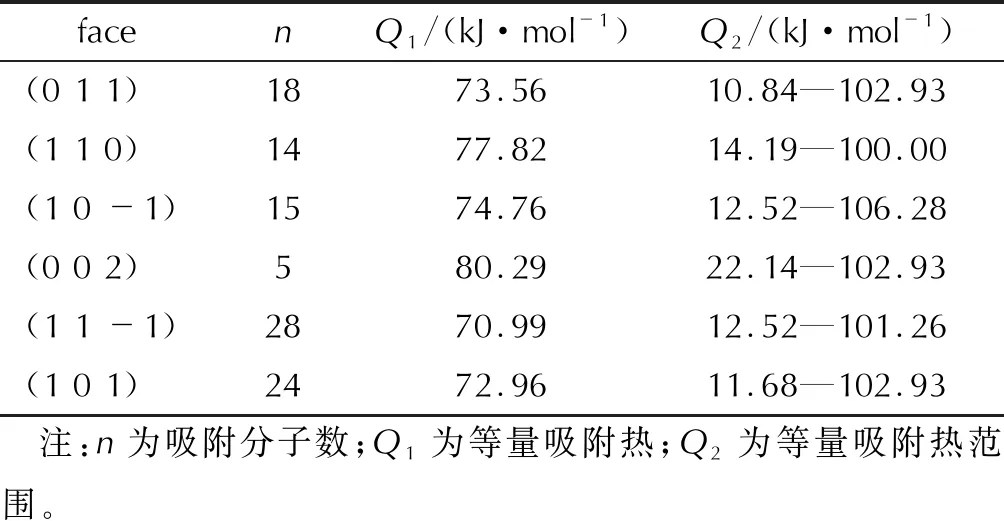
表5 EA在ε-CL-20不同晶面上吸附的EA分子数和等量吸附热Table 5 Number of adsorbed EA molecules and isosteric adsorption heat on six faces of ε-CL-20
3 结 论
(1)AE模型预测真空中ε-CL-20有6个晶面,各个晶面生长速率的大小顺序为:(1 0 1)>(1 1 -1)>(0 0 2)>(1 1 0)>(1 0 -1)>(0 1 1),粗糙度大小顺序为:(1 0 -1)>(1 1 -1)>(0 1 1)>(1 0 1)>(1 1 0)>(0 0 2) 及带隙大小顺序为:(0 0 2)>(1 0 1)>(0 1 1)>(1 0 -1)>(1 1 -1)>(1 1 0)。
(2)ε-CL-20晶面的活性位点主要位于台阶、扭折或空隙处,EA分子通过C—H…O弱氢键、范德华力等相互作用进入活性位点,与晶面形成一定厚度的溶剂界面层。其中,EA分子通过(1 1 -1)面的袋状空隙进入晶格,形成最厚的界面溶剂层(约10Å),而EA分子只能吸附在(0 0 2)平坦的表面上,形成最薄的界面溶剂层(约4Å),其他4个晶面介于这两个晶面之间。溶剂层与晶面的结合能大小顺序为:(1 1 -1)>(1 1 0)>(1 0 -1)>(1 0 1)>(0 1 1)>(0 0 2)。
(3)单个EA分子在不同晶面上的吸附能绝对值大小顺序为:(1 1 -1)>(1 0 -1)>(1 1 0)>(1 0 1)>(0 1 1)>(0 0 2),吸附的EA分子数为:(1 1 -1)>(1 0 1)>(0 1 1)>(1 0 -1)>(1 1 0)>(0 0 2) 及等量吸附热为:(0 0 2)>(1 1 0)>(1 0 -1)>(0 1 1)>(1 0 1)>(1 1 -1)。正的吸附热和负的吸附能值表明EA分子在不同晶面上的吸附均为放热过程。
(4)基于校正后的附着能及吸附能预测的晶面生长速率基本一致,即生长最快的为(0 0 2)面,最慢的为(1 1 -1),其次是(1 0 -1),其他3个晶面的生长速率介于其间。(1 1 -1)面在形态学上的重要性将增加,而(0 0 2)在最终形态中的显露面将减小甚至消失。
