基于Csy4与MCP的新型迷你基因组编辑系统的构建
2023-11-23邓嘉辉雷建峰赵燚刘敏胡子曜尤扬子邵武奎柳建飞刘晓东
邓嘉辉 雷建峰 赵燚 刘敏 胡子曜 尤扬子 邵武奎柳建飞 刘晓东
(1. 新疆农业大学生命科学学院,乌鲁木齐 830052;2. 新疆农业大学农学院 教育部棉花工程研究中心 农业生物技术重点实验室,乌鲁木齐 830052)
基因组编辑技术是指利用人工构建的特异性核酸酶对生物体核酸序列进行删除、插入、替换等各种修饰的基因工程技术。该技术的出现为生物体功能基因组学研究提供了便捷的反向遗传学手段[1-2]。目前,基因编辑技术主要包括锌指核酸酶技术(zinc finger nucleases, ZFNs)、类转录激活因子效应物核酸酶技术(transcription activator‑like effector nucleases, TALENs)及CRISPR/Cas(clustered regularly interspaced short palindromic repeat‑associated protein)系统,其中,CRISPR/Cas系统由于编辑效率高、操作耗费时间少和成本低等优点,成为当前广泛应用的主流基因编辑系统[3-4]。
ZFN由锌指蛋白(zinc finger protein, ZFP)和限制性内切酶Fok I融合构成。3-5个串联的ZFP构成ZFN的DNA结合结构域,可与DNA靶位点特异性结合,然后引导FokI蛋白二聚体发挥DNA切割功能[5],之后通过引导核酸内切酶切割DNA靶位点序列,形成DNA双链断裂(DNA double‑strand break, DSB),从而诱导细胞进行DNA损伤修复。细胞通过非同源末端连接(non‑homologous end joining,NHEJ)或同源重组(homology‑directed repair, HDR)两种不同的DNA修复途径来实现基因组编辑,包括基因敲除(knockout)、基因定点突变及外源基因的定点整合(knockin)等[6]。TALEN技术则继承ZFN技术的设计原理,通过类转录激活效应因子(transcription activator‑like effector, TALE)与DNA靶位点特异结合,诱导FokI蛋白二聚体在靶位点进行切割[7-9]。CRISPR/Cas9技术是通过人工合成的引导RNA(single strand guide RNA, sgRNA)序列与靶位点特异性结合,引导Cas9蛋白进行DNA双链切割[10]。
第一、二代基因组编辑技术ZFNs和TALENs虽然实现了特异性地识别靶位点并对靶标基因进行敲除,但是其构建繁琐、难度大、时间长、脱靶率高、可操作性不高等问题严重限制了它们的应用范围[11]。CRISPR/Cas技术虽然突变效率、精确度及安全性更高,耗费时间更少,但是其靶向核酸酶分子量大、依然存在脱靶现象和受PAM识别序列的限制等问题,尚需要进一步改进[12]。
MCP蛋白(major capsid protein, MCP)是MS2噬菌体的外壳蛋白,由130个氨基酸组成,它可以精确识别并结合由19或者21个核苷酸组成的MS2小RNA茎环结构(MS2‑SL)[13-14]。目前,MCP蛋白被用来识别MS2‑SL茎环结构并融合转录抑制因子或者转录激活因子来控制下游基因的表达[15]。Csy4(CRISPR associated protein, Cas6f)是一种参与CRISPR1‑F系统crRNA生成的RNA酶,由188个氨基酸组成,其发挥功能除了需要识别特异性靶序列外,还需要靶序列3′端形成5个碱基配对的茎环结构。因为同时对碱基序列和二级结构的要求导致Csy4相比传统内切酶具备较高的特异性[16-17]。由于MCP蛋白具有识别并结合guide‑SL的功能,Csy4蛋白也可以识别并结合sgRNA,它们的功能类似于CRISPR/Cas9基因编辑系统内Cas9蛋白,但它们并无切割活性。
本研究基于一代基因编辑技术ZFNs和二代基因编辑技术TALENs的原理,选取分子量小、RNA高结合能力和高特异性的MCP蛋白和Csy4蛋白,与具有核酸内切酶特性的FokI蛋白构建融合蛋白。拟建立MCP‑FokI和Csy4‑FokI新型双靶位点编辑系统,将FokI蛋白构建至靶向蛋白MCP或Csy4蛋白的C端和N端,让正反两个靶向蛋白分别识别目标基因位点左右两侧的DNA序列,通过探究不同的中间间隔区的间距使FokI在靶位点形成活性二聚体,进而发挥内切酶活性,造成DSB,细胞通过HDR或NHEJ的机制进行修复进而实现基因编辑(图1)。希望通过构建MCP‑FokI和Csy4‑FokI这两个新型双靶位点编辑系统来解决CRISPR/Cas基因组编辑技术存在的脱靶现象和受PAM识别序列限制等问题。
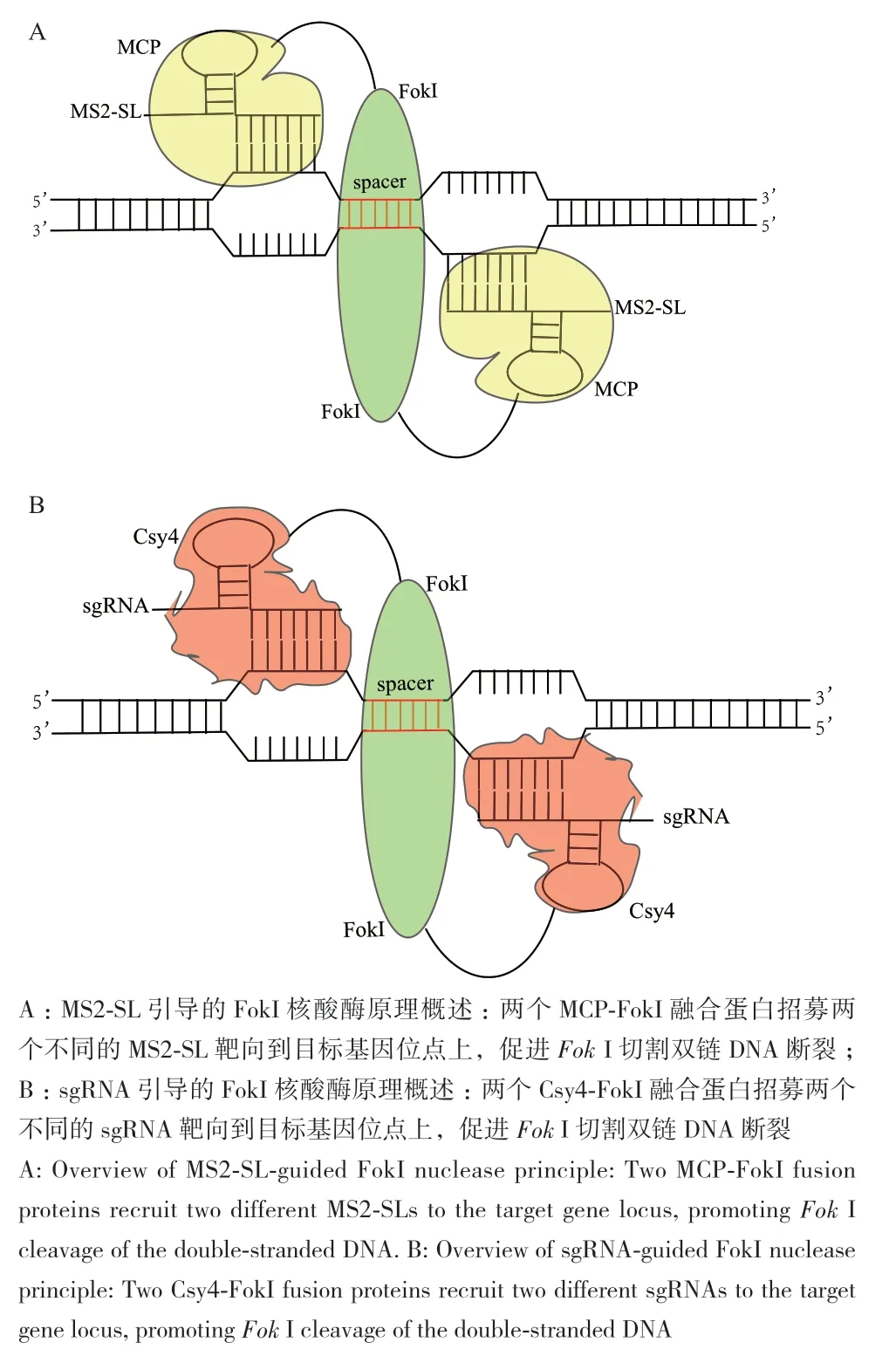
图1 MCP-FokI(FokI-MCP)和Csy4-FokI(FokI-Csy4)基因组编辑体系示意图Fig. 1 Schematic diagram of MCP-FokI(FokI-MCP)and Csy4-FokI(FokI-Csy4)genome editing system
1 材料与方法
1.1 材料
所用AtU6‑26∷sgRNA载体、棉花叶皱缩病毒载体CLCrV‑A和CLCrV‑B均为新疆农业大学作物功能基因组学实验室所保存,载体CLCrV‑A和CLCrV‑B由浙江大学周雪平教授惠赠。植物基因组DNA提取试剂盒(EasyPure Plant Genomic DNA Kit)、克隆载体(pEASY‑B Zero)和感受态细胞(Trans1‑T1)均购于北京全式金生物公司。所用限制性内切酶购于赛默飞生物公司。引物合成及测序由上海生工生物科技有限公司完成,其余试剂及药品均由新疆本地生物公司代购。
1.2 方法
1.2.1 迷你基因组编辑系统不同中间间隔区的设计 分别使用2个不同U6启动子驱动成对的guide‑SL左侧靶位点和右侧靶位点,以减少使用相同启动子导致基因沉默的概率。已有2个单独研究团队研究表明CRISPR/dCas9‑FokI对EGFP编辑效率最高的中间间隔区的距离为14-25 bp[18]、13-17 bp[19]。因为并不知道有效切割所需的间隔距离,因此本研究设计4对针对AtCLA1不同位点的guide‑SL和7对不同位点的sgRNA来探究迷你基因组编辑系统的编辑能力。
1.2.2 MCP‑FokI(FokI‑MCP)和Csy4‑FokI(FokI‑Cs‑y4)基因编辑载体的构建 参照雷建峰[20]方法构建基因编辑载体,首先构建35∷MCP‑FokI‑Ter‑B zero和35∷FokI‑ MCP‑Ter‑B zero过渡载体,并经Xho I、Sal I、Xba I和BamH I酶切鉴定。构建4个AtU6∷SL‑B zero和4个AtU6‑26∷SL‑B zero过渡载体,经Kpn I和BamH I双酶切鉴定。将构建好的靶向敲除AtCLA1的不同AtU6‑26∷SL和AtU6∷SL片段与35∷MCP‑FokI‑Ter和35∷FokI‑MCP‑Ter片段分别重组为35∷MCP‑FokI‑Ter‑P1300和35∷FokI‑MCP‑Ter‑P1300载体,经Hind III、Sal I、Xba I和Kpn I酶切鉴定,验证是否成功。
通过Transfer‑PCR目的片段替换的方法[21]对35S∷MCP‑FokI‑Ter和35S∷FokI‑MCP‑Ter表达载体进行改造,将MCP片段替换为Csy4,经Dpn I酶切3 h,转化、测序,并经Spe I和Pac I酶切鉴定。分别在35S∷FokI‑Csy4载体与35S∷Csy4‑FokI载体的5′端添加Spe I酶切位点,3′端添加Pac I酶切位点,经测序比对和Spe I与Pac I双酶切鉴定,成功构建Spe I‑35S∷Csy4‑FokI‑B zero‑Pac I和Spe I‑35S∷FokI‑Csy4‑B zero‑Pac I载体。设计7对针对CLA1不同位点的sgRNA,分别对4个AtU6∷SL‑B zero和2个AtU6‑26∷SL‑B zero载体进行改造,通过Tansfer‑PCR目的片段替换的方法将SL片段替换为Csy4‑sgRNA,构建AtU6∷sgRNA(1-4)‑B zero和AtU6‑26∷sgRNA(5-6)‑B zero 6个过渡载体,分别在这6个载体的5′端添加Pac I酶切位点,3′端添加Avr II酶切位点,之后将4个AtU6∷sgRNA和2个AtU6‑26∷sgRNA片段两两串联连接到SK载体上,经Kpn I、EcoR I和Xba I酶切鉴定。将FokI‑Csy4和Csy4‑FokI片段与CLCrV‑A载体连接,同时,将其分别与7个不同间距的双靶序列片段与CLCrV‑A载体串联连接,将FokI‑Csy4和Csy4‑FokI与CLCrV‑A载体连接,同时将其分别与7个不同间距的双靶序列片段与CLCrV‑A载体串联连接,经Spe I、Avr II双酶切鉴定靶向敲除拟南芥AtCLA1的Csy4‑FokI和FokI‑Csy4编辑载体是否构建成功。
1.2.3 转化农杆菌感受态 吸取重组成功的CLCrV‑A基因编辑载体与CLCrV‑B质粒各20 μL,加入100 μL刚刚解冻的农杆菌GV3101感受态中,冰浴35 min,液氮速冻2 min,立即放入37℃水浴锅中热激3 min,加600 μL无抗LB液体培养基,28℃、220 r/min恒温摇床中孵育4 h,在LB固体培养基(含50 μg/mL Rif和50 μg/mL Kan)上筛选阳性克隆,酶切鉴定,得到含编辑载体的阳性转化菌用于注射拟南芥叶片。
1.2.4 浸花法转化拟南芥 采用浸花法[22]对野生型拟南芥Col‑0进行转化。取农杆菌冻存液10 μL接种于2 mL LB培养基(50 μg/mL Kan和50 μg/mL Rif)中活化。28℃,200 r/min振荡培养20 h左右。然后按1∶100体积比将活化菌液接种到25 mL LB培养基(50 μg/mL Kan和50 μg/mL Rif)中,于28℃,200 r/min振荡培养,摇至菌液的OD600≈1.6-1.8时,离心收集农杆菌培养物,重悬于新鲜配制的转化液(1/2 MS液体培养基含5%蔗糖,0.02% Silwet L‑77),至终浓度OD600≈0.8-1.0,转化拟南芥Col‑0。在筛选转化子时,种子用5% NaClO消毒5 min,置于1/2 MS培养基(含30 μg/mL Kan)上萌发。2周后,将阳性苗移栽到土壤中,在22℃ 16 h光照/8 h黑暗条件下生长。选取生长15 d左右的拟南芥幼苗进行接种转化试验。
1.2.5 瞬时转化拟南芥 将‑80℃保存的阳性转化菌在LB固体培养基(含50 μg/mL Rif和50 μg/mL Kan)上划线活化,挑取单克隆接种于体积为10 mL LB液体培养基(含50 μg/mL Rif和50 μg/mL Kan)中,28℃、220 r/min恒温摇床中摇至OD600≈1.5左右,4 000 r/min离心10 min沉淀菌体,接着用含有10 mmol/L MgCl2、10 mmol/L MES和200 μmol/L乙酰丁香酮的悬浮液悬浮至OD600≈1.0,室温遮光静置3 h。接种时,将CLCrV‑A和CLCrV‑B悬浮菌液按1∶1等量混合,使用1 mL注射器注射到拟南芥叶片中进行瞬时表达。
1.2.6 突变检测 剪取注射农杆菌后的拟南芥叶片100 mg左右,提取基因组DNA。参照Gao等[23]方法检测AtCLA1是否发生编辑。利用PCR扩增出含有EcoR V和Xba I酶切位点的目的片段(表1)。用对应的限制性内切酶消化扩增产物,检查是否存在未被消化的PCR扩增条带,初步判定是否发生基因编辑,通过HI‑TOM高通量测序,将测序结果与基因序列进行比对,确定AtCLA1的突变类型。

表1 引物及用途Table 1 Primers and applications
1.2.7 融合蛋白结构预测 利用蛋白结构数据库AlphaFold2(https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/ AlphaFold2.ipynb)对融合蛋白的结构进行预测。
1.2.8 瞬时表达载体检测 以注射7个不同中间间隔区距靶序列的Csy4‑FokI基因编辑载体14 d后的子叶叶片基因组DNA为模板,分别以未注射农杆菌的拟南芥Col‑0和注射未带靶序列的Csy4‑FokI载体为阴性对照。利用CLCrVB病毒载体上特异性引物(表1)进行PCR扩增。
2 结果
2.1 融合蛋白结构分析
由于MCP和Csy4蛋白较小,与FokI融合后是否会影响彼此的三维空间,进而影响各自功能的发挥。运用AlphaFold2蛋白结构数据库对MCP‑FokI和FokI‑MCP融合蛋白进行预测(图2‑A),将FokI蛋白和MCP蛋白的预测结果图同2个融合蛋白的预测结果图进行比对,2个功能域融合后,并没有改变FokI蛋白形成二聚体的功能结构,也没有改变MCP蛋白识别并结合guide‑SL的功能结构。通过对Csy4‑FokI和FokI‑Csy4融合蛋白进行预测(图2‑B),并将FokI蛋白和Csy4蛋白的预测结构图同2个融合蛋白的预测结构图进行比对发现,没有改变FokI蛋白形成二聚体的功能结构,也没有改变Csy4蛋白识别并结合sgRNA的功能结构。
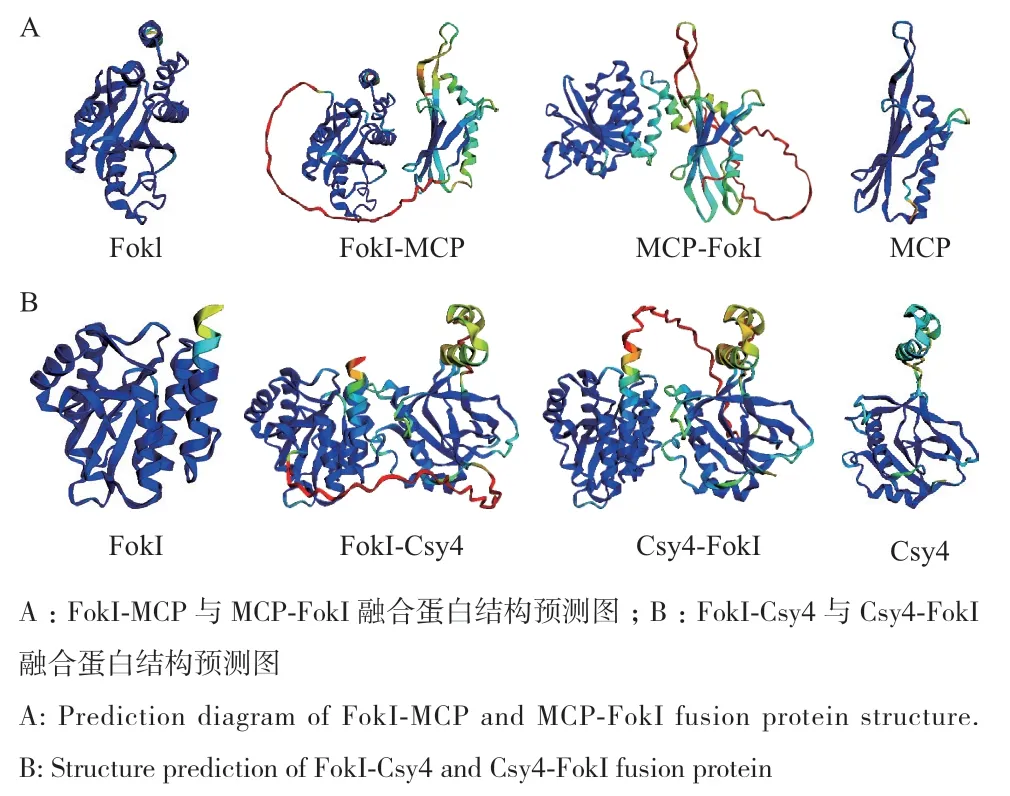
图2 融合蛋白三级结构预测Fig. 2 Tertiary structure prediction of fusion protein
2.2 MCP‑FokI编辑载体构建
本研究设计4对针对CLA1不同位点的guide‑SL,中间间隔区的间距分别为10、14、16和25 bp,中间间隔区的序列上有EcoR V和Xba I酶切位点,便于突变检测(图3‑A)。35∷MCP‑FokI‑Ter‑B zero和35∷FokI‑MCP‑ Ter‑B zero过渡载体(图3‑C)、4个AtU6∷SL‑B zero和4个AtU6‑26∷SL‑B zero靶序列过渡载体(图3‑D)以及带有4个不同中间间隔区的35∷MCP‑FokI‑Ter‑P1300和35∷FokI‑MCP‑Ter‑P1300载体编辑载体(图3‑B, E)经酶切鉴定及测序比对后与预期结果相吻合,说明靶向敲除AtCLA1的MCP‑FokI和FokI‑MCP编辑载体已构建成功。
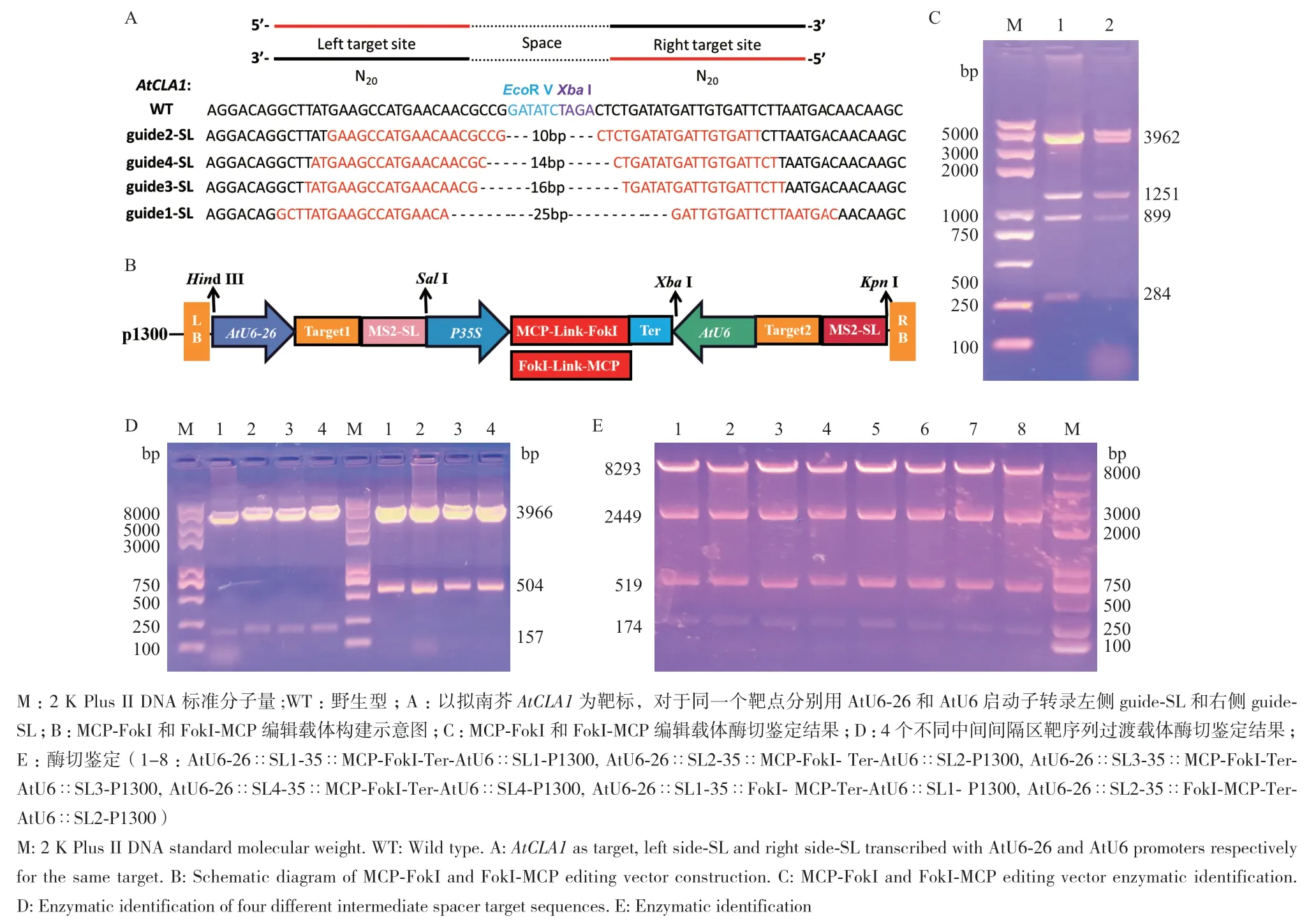
图3 MCP-FokI和FokI-MCP基因编辑载体构建图Fig. 3 Construction of MCP-FokI and FokI-MCP gene editing vector
2.3 MCP‑FokI基因组编辑技术可行性检测
运用浸花法将构建的编辑载体分别转化野生型拟南芥Col‑0,潮霉素筛选获得大量转基因T1代植株(图4)。选取转基因阳性苗基因组DNA为模板进行扩增,PCR扩增覆盖突变位点的AtCLA1。通过Hi‑TOM高通量测序,结果显示,选取的4对不同中间间隔区的CLA1‑SL、MCP‑FokI和FokI‑MCP编辑系统均未能实现对靶基因的靶向编辑,与此同时,并没有观察到AtCLA1突变植株明显的表型。
图4 MCP-FokI和FokI-MCP基因编辑系统靶向敲除AtCLA1Fig. 4 Targeted knockout of AtCLA1 gene by MCP-FokI and FokI-MCP gene editing system
2.4 Csy4‑FokI编辑载体的构建
Spe I‑35S∷Csy4‑FokI‑B zero‑Pac I和Spe I‑35S∷‑FokI‑Csy4‑B zero‑Pac I过渡载体(图5‑C)、4个AtU6∷sgRNA‑B zero和2个AtU6‑26∷sgRNA‑B zero过渡载体、7个中间间隔区的靶序列过渡载体(图5‑D)以及带有7个中间间隔区靶序列的FokI‑Csy4‑CLCrV‑A和Csy4‑FokI‑CLCrV‑A载体(图5‑B, E),经酶切鉴定及测序比对后,与预期结果相吻合,表明说明靶向敲除拟南芥CLA1的Csy4‑FokI和FokI‑Csy4编辑载体已构建成功。

图5 Csy4-FokI和FokI-Csy4基因编辑载体构建图Fig. 5 Construction of Csy4-FokI and FokI-Csy4 gene editing vector
2.5 Csy4‑FokI基因组编辑技术可行性检测
通过叶片注射法将构建的编辑载体,分别注射到野生型拟南芥Col‑0叶片中进行瞬时表达。接种病毒7-14 d后并未观察到拟南芥CLA1被编辑后明显的白化表型。
分别提取野生型Col‑0拟南芥基因组DNA与注射Csy4‑FokI编辑载体的拟南芥基因组DNA,对CLCrV‑B基因组进行PCR扩增。结果显示,在注射Csy4‑FokI编辑载体的拟南芥基叶片中可以检测到CLCrV‑B病毒积累,而在野生型拟南芥未检测到CLCrV‑B病毒的积累(图6‑A, B)。表明编辑载体已成功转入拟南芥中。
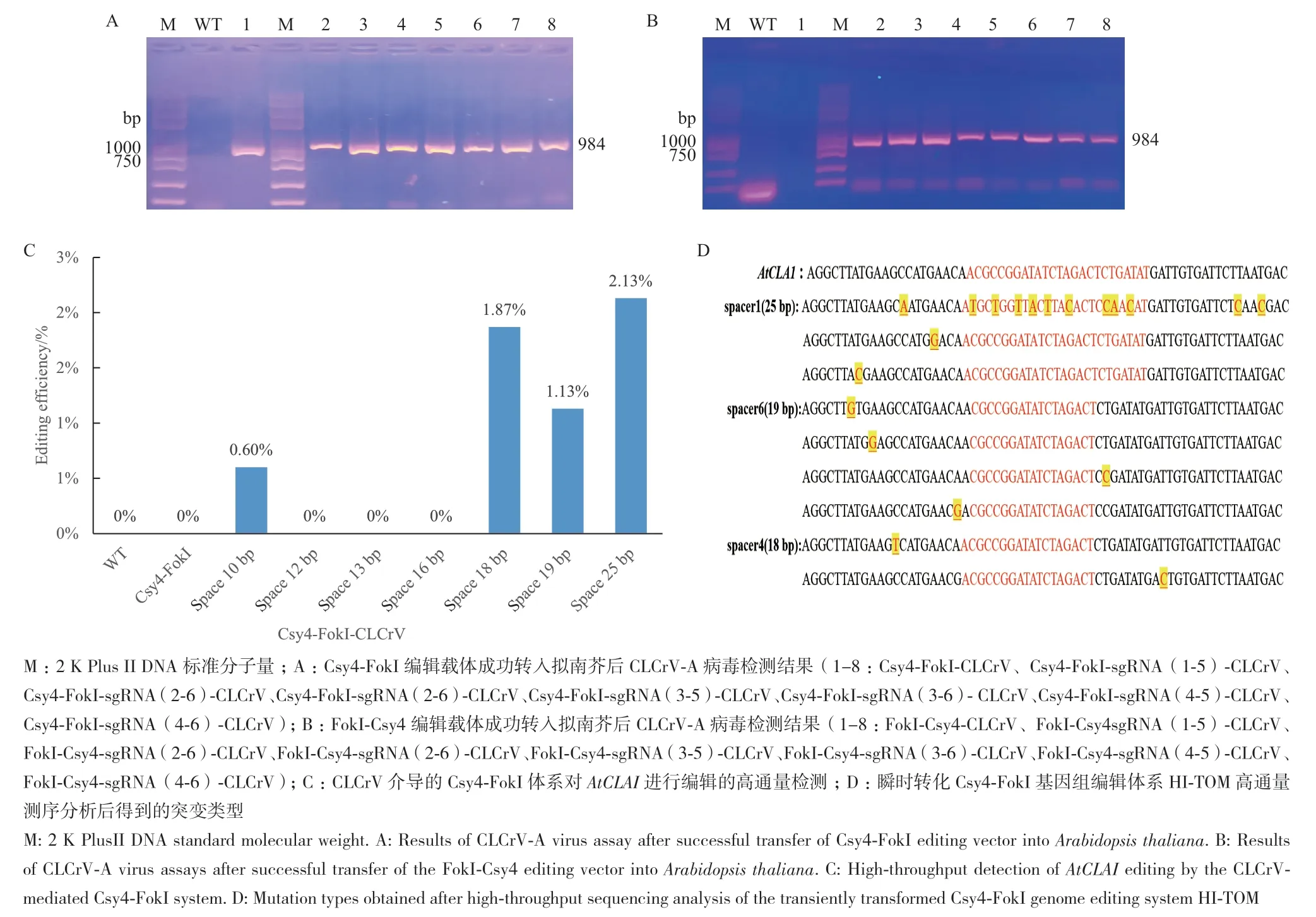
图6 Csy4-FokI和FokI-Csy4基因编辑系统靶向敲除AtCLA1Fig. 6 Targeted knockout of AtCLA1 gene by Csy4-FokI and FokI-Csy4 gene editing system
通过Hi‑TOM高通量测序,结果显示,在Csy4‑FokI基因组编辑体系中,检测范围在靶位点sgRNA(1-5)之间64 bp区域,中间间隔区为25 bp的植株#5、#6和#7出现了3种类型的碱基替换,平均编辑效率为2.13%;检测范围在靶位点sgRNA(2-6)之间48 bp区域,中间间隔区为10 bp的植株#15出现了1种类型的碱基替换,编辑效率为0.3%;检测范围在靶位点sgRNA(1-6)之间57 bp区域,中间间隔区为19 bp的植株#17和#19出现了3种类型的碱基替换,平均编辑效率为1.13%;检测范围在靶位点sgRNA(4-5)之间56 bp区域,中间间隔区为18 bp的植株#5、#6和#7出现了3种类型的碱基替换,平均编辑效率为1.87%。而在野生型、Csy4‑FokI靶位点空载对照和FokI‑Csy4基因组编辑体系中未检测到编辑的发生。结果表明,在Csy4‑FokI基因组编辑体系中选取的7对不同中间间隔区的sgRNA,CLCrV介导的Csy4‑FokI编辑系统中间间隔区为10、18、19和25 bp能够实现对靶基因的靶向编辑(图6‑C),但是突变类型均为碱基置换类型且编辑效率很低,而CLCrV介导的FokI‑Csy4基因组编辑体系和Csy4‑FokI编辑系统中间间隔区为12、13和16 bp均不能实现基因编辑。
3 讨论
基因组编辑技术以准确、高效、简单地操作而发展迅速,近年来,以CRISPR/Cas系统为代表的第三代基因组编辑技术在基因功能研究、植物分子育种、疾病治疗等方面取得了重要进展,但CRISPR/Cas系列基因组编辑技术的应用领域还存在其靶向核酸酶分子量大、存在脱靶现象和受PAM识别序列的限制等问题,研究者一直致力于寻找解决方法,通过改造修饰Cas9蛋白、使用Cas9直系同源酶等一系列措施,来提高基因编辑效率,降低其脱靶率,减弱PAM位点限制来进一步拓展应用范围。本研究构建的MCP‑FokI和Csy4‑FokI融合蛋白可能比Cas9蛋白更具有优势。主要表现在:首先Cas系列基因相对较大,大于4 000 bp[19],而MCP‑FokI和Csy4‑FokI大小为1 074和1 248 bp,从碱基片段大小来讲,两个融合蛋白小于Cas蛋白,更利于编辑载体的构建。此外,Cas对靶位点的切割依赖于PAM位点,尽管经过人工改造的Cas9可以识别更多的PAM位点[24],但是Cas9蛋白发挥切割作用仍然存在PAM位点的限制,导致并不是所有基因任意区段都能被编辑。使用上述融合蛋白靶向目标基因并没有PAM位点的限制,从而提高了目标位置选择的灵活性;最后,也就是说CRISPR/Cas9并没有实质性地解决脱靶问题[25]。若将两个融合蛋白应用于基因编辑体系必须采用双切口酶策略,也就是用2个不同的U6启动子分别驱动guide‑SL或sgRNA的转录,Fok I分别切割目标DNA片段的正负链,产生DNA双链断裂。只有2个guide‑SL或sgRNA同时靶向到同一个基因上,才能发挥基因编辑的功能,从而提高了基因编辑的特异性进而解决CRISPR/Cas9的脱靶问题。本实验构建的新型迷你基因组编辑系统虽然具有蛋白小、不受PAM位点限制和脱靶效率低等优点,但仍存在一些限制。该系统是一种双靶位点编辑系统,需要在目标基因的两侧设计靶向位点,因此,在设计和载体构建上具有一定的复杂性。此外,迷你基因组编辑系统也会受到靶向蛋白自身性质的影响,例如,与RNA结合能力的强弱、RNA切割活性和靶向蛋白的大小等,这些因素都可能影响该系统的编辑能力。
根据报道[26-27]在CRISPR/dCas9‑FokI编辑体系内,FokI在dCas9蛋白的N端,通过设计有效的靶序列才可以对目标基因进行编辑。出现这样结果的原因可能是dCas9和FokI这两个蛋白在空间构象上有严格的排列顺序。为此,本研究构建了4种不同的融合蛋白:FokI核酸酶域与MCP(MCP‑FokI)的羧基末端融合和与其氨基末端融合(FokI‑MCP),FokI核酸酶域与Csy4(Csy4‑FokI)的羧基末端融合和与其氨基末端融合(FokI‑Csy4),融合蛋白在结构上与ZFNs和TALENs相似(图7)。这4种融合蛋白本研究使用带有SGGSSGGSSGSETPGTSESAT‑PESSGGSSGGS序列的32个氨基酸连接子来连接这两个结构域,同时对MCP蛋白进行了密码子优化,使其能在植物细胞中表达[20]。
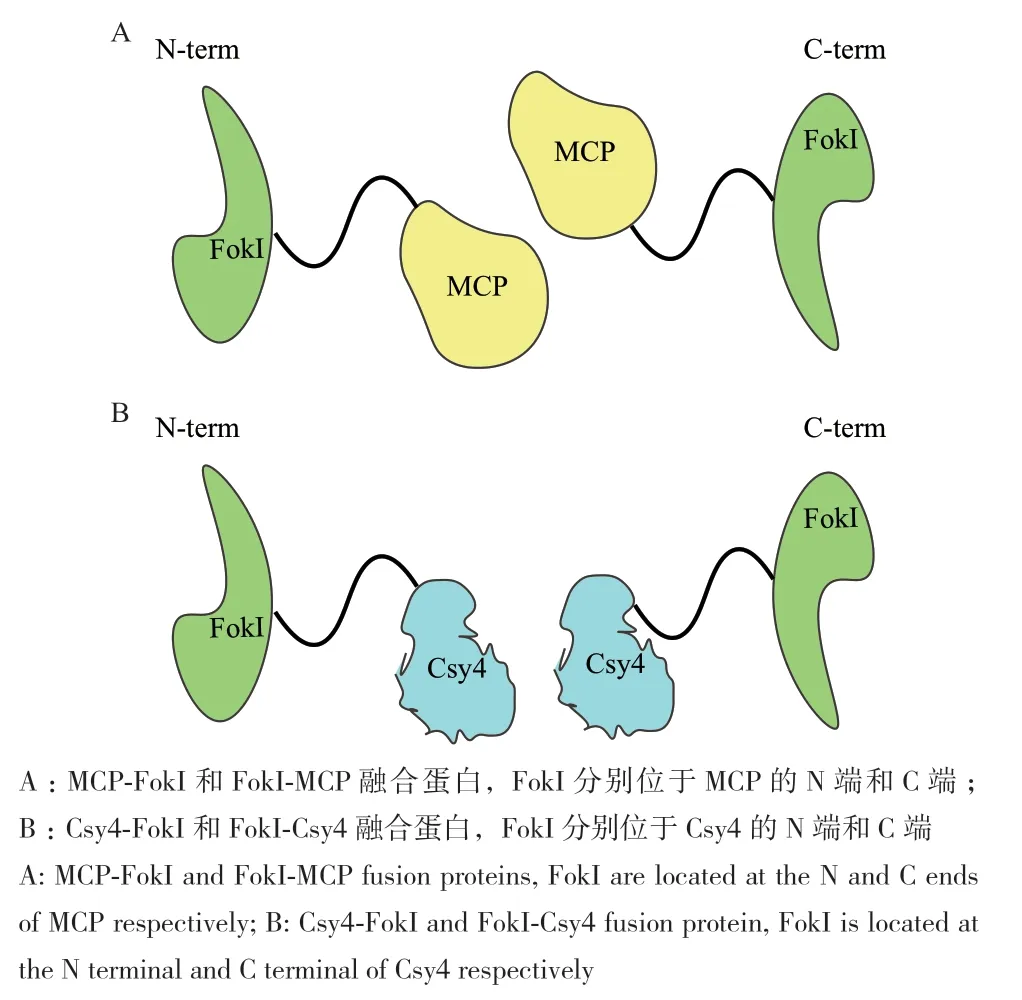
图7 MCP-FokI(FokI-MCP)和Csy4-FokI(FokI-Csy4)融合蛋白示意图Fig. 7 Schematic diagram of MCP-FokI(FokI-MCP)and Csy4-FokI(FokI-Csy4)fusion protein
病毒诱导的基因编辑(virus‑induced genome editing, VIGE)技术能够改造植物病毒使其能在不同植物中表达外源蛋白和特定长度的RNA,可以通过简单的转化方法借助病毒在植物体内系统扩散能力将基因组编辑元件投送到植物细胞内[28],提高了基因编辑的检出率。棉花叶皱缩病毒(cotton leaf crumple virus, CLCrV)是一种靠烟粉虱传播的双生病毒,由CLCrV‑A和CLCrV‑B两个环状单链DNA成分组分构成[29]。Gu等[30]改良了基于CLCrV病毒诱导的基因沉默(virus‑induced gene silencing, VIGS)载体,在棉花中实现了高效持久的基因沉默。Lei等[31]基于CLCrV介导的VIGE体系,在拟南芥中利用FT‑sgRNA策略成功获得了可遗传的基因编辑后代。赵燚等[32]采用CLCrV介导VIGE体系,成功编辑了棉花内源基因。利用病毒载体进行基因编辑已在多种植物中得到成功应用,但是受病毒基因组承载外源DNA片段的限制阻碍了其承载大片段DNA进行基因编辑的应用。然而也有研究报道少数病毒可承载完整的基因编辑载体进行基因编辑,如Ma等[33]采用苦苣菜黄网病毒(sonchus yellow net virus, SYNV)将整个CRISPR/Cas9系统投递到烟草体内的,并实现了基因编辑;Ariga等[34]利用马铃薯病毒X(potato virus X, PVX)病毒载体表达Cas9和sgRNA成功在本氏烟草中实现了高效编辑;Liu等[35]基于一种广寄主范围的番茄斑萎病毒(tomato spotted wilt virus, TSWV)建立了高效的遗传操作系统。研究者通过建立瞬时递送系统向多种作物不同品种稳定递送一系列大的CRISPR/Cas核酸酶分子,展现出SYNV、PVX和TSWV具有较好的承载力。CLCrV能否像以上病毒一样能够承载完整的编辑系统,进而快速验证Csy4‑FokI基因组编辑系统的编辑能力。为此,本研究尝试将完整的编辑体系构建到CLCrV病毒载体上,利用CLCrV病毒载体投送到拟南芥叶片中快速验证Csy4‑FokI基因编辑体系的可行性。
本研究构建的MCP‑FokI基因组编辑系统通过稳定遗传转化拟南芥的方式并未检测到基因编辑的发生,推测其原因可能有:设计的sgRNA不同中间间隔区的设计是否有效、蛋白融合方式、与RNA结合能力的强弱。其中需要解决的是通过蛋白质与RNA互作检测手段来明确MCP蛋白与MS2‑guide‑SL的结合能力。如果结合能力较弱,可以通过串联更多的MS2‑SL以提高MCP蛋白与MS2‑guide‑SL的结合能力,进而提高编辑效率。
对于Csy4‑FokI编辑系统,利用CLCrV病毒载体投送到拟南芥叶片中来快速验证Csy4‑FokI基因编辑体系。经过Hi‑TOM高通量检测结果的分析发现,CLCrV介导的Csy4‑FokI编辑系统的编辑效率较低。推测造成这一问题的原因可能包括以下几点:首先在该系统中,构建的融合蛋白的基因片段相对较大(3 316 bp)。然而,CLCrV病毒载体在承载了3 000 bp外源基因片段后,可能会导致其复制能力和扩散侵染能力大幅下降,不能侵入更多的组织细胞。由于病毒载体需要有效感染目标细胞才能实现基因编辑,因此低细胞感染率可能是导致编辑效率低的主要原因之一。其次,Csy4‑FokI编辑系统的靶向蛋白除具有和sgRNA高识别与结合特异性外,还存在RNA切割活性。这可能导致sgRNA在与Csy4靶向蛋白结合后被切割,从而降低了结合的能力。此外,使用Csy4蛋白进行植物基因组编辑存在一定的风险,因为研究发现在植物中引入Csy4蛋白加工sgRNA或pegRNA时可能产生毒性[36-37]。这可能是因为Csy4蛋白影响了植物mRNA的功能,对植物细胞产生了毒害作用,进而影响了编辑效率。后续研究将对Csy4蛋白进行改造,使其不具有RNA切割活性,而只是作为RNA结合蛋白来进一步验证该迷你基因组编辑系统的编辑效率。未来的研究也可以尝试通过验证更多基因和设计不同的中间间隔区,并采用浸花法等稳定转化的方式来验证其编辑能力。
综上所述,本研究采用病毒诱导的基因编辑(VIGE)技术注射拟南芥叶片,初步探索了Csy4‑FokI与MCP‑FokI小型基因组编辑体系的编辑能力,为今后优化并利用该技术体系进行植物分子育种奠定前期基础。
4 结论
初步建立了Csy4‑FokI的迷你的、无PAM位点限制的新型基因组编辑系统。CLCrV介导的Csy4‑FokI迷你基因组编辑系统能够实现对目标基因进行靶向编辑,但是检测到的突变类型只有碱基置换,且编辑效率很低。
