DFT 计算在铁基催化剂费托合成反应研究中的应用
2023-11-21何富贵李海鹏何育荣高新华张建利赵天生
何富贵,张 曈,梁 洁,李海鹏,何育荣,*,高新华,2,*,张建利,赵天生
(1.宁夏大学化学化工学院 省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏 银川 750021;2.国家能源集团宁夏煤业有限责任公司煤炭化学工业技术研究院,宁夏 银川 750411)
碳氢化合物在社会发展中被广泛应用,传统生产碳氢化合物的工艺是从煤、石油、天然气等不可再生能源中获得。随着全球人口数量指数级增长,对不可再生能源的需求不断增加,能源消耗的同时大气中COx浓度也在持续上升,带来了一系列的环境问题[1]。在过去的几十年时间里,通过费托合成(Fischer-Tropsch Synthesis,FTS)将CO/CO2转化为具有高附加值的化工原料受到了广泛的关注[2],该合成方法是煤制液体燃料(CTL)工艺的核心技术。使用费托合成反应技术催化转化COx,在有效减缓工业气体对环境造成污染的同时,也有效缓解了当今的能源需求[3]。
FTS 反应由Frans Fischer 和Hans Tropsch 于1923 年提出[4],如图1 所示,该反应用合成气或CO2与H2的混合气作为原料。费托催化剂中使用的典型活性金属是Fe、Co、Ni 和贵金属等[5]。其中,贵金属以Ru 为代表。铁基催化剂的活性相为FeCx,其余均以其零价单质为活性相[6]。Ru 是最活跃的CO 加氢催化剂,能够在低温下催化合成长链碳氢化合物。Ru 还可以在没有任何助剂的情况下高效工作,因此,在探究催化剂的作用机制和反应机理方面Ru 基催化剂能提供最直接的基础认识。然而,高昂的成本和有限的储量阻碍了其工业化应用[7]。Ni 基催化剂具有优异的甲烷化性能,但其活性相在反应过程中易于生成羰基镍从反应器中挥发流失,导致Ni 基催化剂难以实现工业化应用[8]。

图1 氧化铁和碳化物组成的双官能活性位点上CO2 加氢生成碳氢化合物的示意图[9]Figure 1 Scheme of CO2 hydrogenation to hydrocarbons at bifunctional active sites composed of iron oxide and carbide[9](with permission from ACS publications)
Fe、Co 基催化剂已被用作工业化费托催化剂,Co 基催化剂通常对线性长链烃类具有更强的活性和选择性,且不易被水钝化。因此,Co 基催化剂在合成长链碳氢化合物(如石蜡和柴油)方面受到了广泛关注。南非的萨索公司经过60 年的发展,实现了Co 基催化剂煤制油的工业化生产,满足了南非燃油市场28%的需求[10]。但Co 基催化剂价格偏高,抗硫等毒物性能差,适应温度范围较窄。相比之下,Fe 基催化剂可以在更大温度范围和H2/CO 比下工作且不会提高CH4的选择性。铁基催化剂比Co 或Ru 基催化剂表现出更高的逆水煤气变换(Reverse Water Gas Shift,RWGS)反应活性。这更适用于从煤或生物质中获得的较低H2/CO比率的合成气的转化。这些优点使得铁基催化剂在煤制液体燃料(CTL)或生物质制液体燃料(BTL)技术以及合成气制烯烃过程中具有很大的吸引力[11,12]。
如图2 所示,催化实验在空间尺度上是宏观的,在时间尺度上是秒级或分钟级的,缺乏在皮秒、飞秒级以及原子尺度的探索,由于多相催化的复杂性,要将宏观的实验同微观尺度的变化连接起来非常的困难。在过去几十年的时间,通过原位表征技术解释微观反应机理取得了巨大的进展。但这对实验条件和设备提出较高的要求,且实验条件下真实反应的复杂性使得难以对单一要素所起的作用进行定性分析。在这种情况下,通过理论计算的方法,在特定条件下的反应模型上进行理论计算,建立了微观尺度与宏观尺度之间的桥梁。
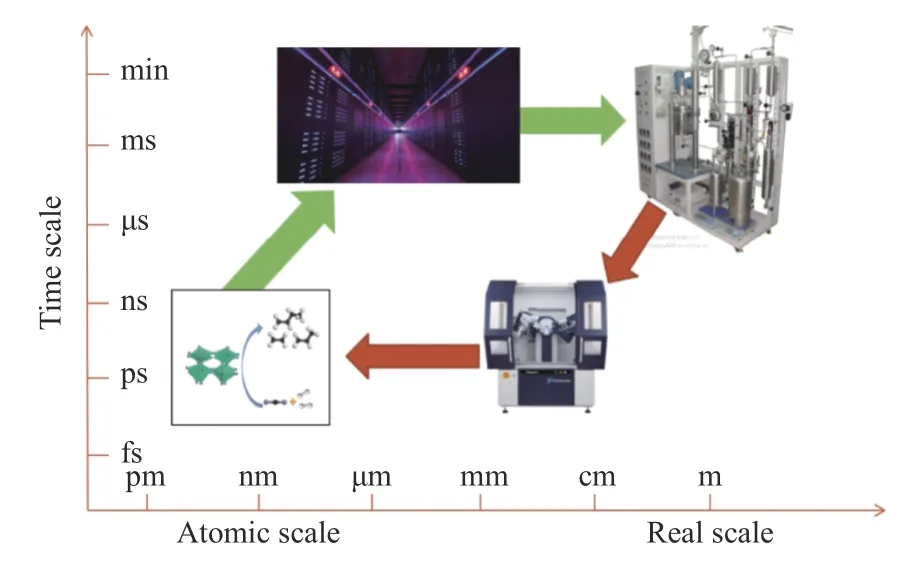
图2 理论计算、原位表征、催化实验在时间和空间尺度对比示意图Figure 2 Comparison of theoretical calculation,in-situ characterization and experimental at time and space scales
针对铁基费托催化,越来越多的实验研究证明了碳化铁物相在FTS 反应中起着重要的作用[13]。要想了解FTS 反应的转化机理,确定反应过程中催化剂不同中间相的具体作用是指导设计催化剂的关键。由于铁碳化物相的形成受碳化学势、温度和压力等因素的影响,不同铁碳化物相间存在复杂的动态转变,因此,实验条件下难以获得绝对纯净的单相碳化铁[14]。杂质效应结合可能的相变也使“单相”实验研究的论点不那么吸引人[15]。碳化铁的表面形貌重构已经被先进的表面化学研究明确证实,并导致表面组成和取向发生动态变化和组合[16]。因此,从实验研究中得出纯物相反应效果的结论仍然具有挑战性。考虑到上述问题,用理论化学策略来阐明基于FTS 中特定碳化铁相模型催化剂的内在面相关工作原理,作为对目前通过实验方法建立认识的补充已成为一种重要方法。本工作针对密度泛函理论(DFT)计算在铁基FTS反应中的应用以及研究进展进行了分析和概述,并对其进行了展望,以期能提供一些关于铁基FTS 催化反应的基础见解。
1 铁碳化合物的表面化学性质
1.1 铁基催化剂物相
1.1.1 费托反应下的物相组成
在反应过程中,铁基催化剂存在复杂的动态演变,同时包含铁氧化物相(Fe2O3、Fe3O4、FeO)、金属Fe(α-Fe)相和铁碳化物相(ε-Fe2C、ε′-Fe2.2C、χ-Fe2.5C、Fe7C3和θ-Fe3C)[17]等多种物相。这些物相以Fe3O4为核心,表面由FeO、Fe 和碳化铁共同组成,构成了铁基催化剂的工况状态[18,19]。总结已有的研究,不同的反应条件导致不同中间相的形成。氧化铁首先从α-Fe2O3转变为Fe3O4,这与预处理使用的活化气体无关[20]。Fe3O4在不同的活化氛围中发生不同的转变。(1) 在H2气氛下活化时,发生Fe2O3→ Fe3O4→ FeO → Fe 反应,高温和提高H2体积分数有利于还原反应的发生,反应由表相向体相依次进行[21,22];(2) 在CO 气氛下活化时,发生α-Fe2O3→ Fe3O4+ε-Fe2C+ε′-Fe2.2C(CO 体积分数 >20%) → Fe3O4+χ-Fe2.5C(表面相,CO 体积分数 >20%) → FeO(内体相)+θ-Fe3C(外体相,CO 体积分数 >20%)→ Fe(表面相) → Fe/C(铁-碳合金,表面相,CO 体积分数 >40%)反应,高温有利于还原反应的发生,提高CO 体积分数有利于FexC 的生成(当CO 体积分数 <20%时,ε-Fe2C 和ε′-Fe2.2C 含量低于检测下限),反应由表相向体相依次进行[23,24];(3) 在H2+CO 气氛下活化时,反应式与CO 气氛活化时相同,高温有利于还原反应的发生,提高CO 体积分数有利于FexC 的生成,但H2的存在有利于χ-Fe2.5C 的生成,反应同样由表相向体相依次进行[24,25]。
铁氧化物具有多种不同的物相,如α-Fe2O3、γ-Fe2O3、FeO、Fe3O4、α-FeOOH、γ-FeOOH 等,它们各自在FTS 转化中起着不同的作用[26,27]。Fe3O4是RWGS 反应的活性相[28],RWGS 反应是吸热反应,通常需要较高的温度(>300 ℃),因此,RWGS 反应过程中α-Fe2O3、γ-Fe2O3、Fe3O4相可以同时存在[29],FeO 是RWGS 反应的失活相,FeOOH 通常用于电催化或环境催化[30]。
α-Fe 上可以发生CO 的解离和加氢,*CO 在纯净铁表面具有更低的直接解离势能[31,32],在FTS 反应过程中可以分解成*C 和*O,这些*C 物种扩散到α-Fe 晶格中形成铁碳化物相[33]。不同的铁碳化物在FTS 反应中起着重要的作用。关于这些相中哪些控制铁基催化剂的活性和选择性一直存在争议。由于各种物相具有不同的表面电子结构状态,展示出的催化性能具有显著差异[34],因此,了解不同碳化铁物相的表面结构/电子性质以及它们的催化性能及机制至关重要。
铁碳化物归类为间质碳化物,铁碳化物依据*C 物种扩散到α-Fe 晶格原子的三棱柱或者八面体间隙位置形成的不同结构而被依次分为三棱柱状碳化物(TP)和八面体碳化物(O),实验研究观察到的几种稳定存在的铁碳化合物相有Fe7C3、χ-Fe5C2、θ-Fe3C、ε'-Fe2.2C 和ε-Fe2C 相。其中,“TP 碳化物”有Fe7C3,θ-Fe3C 和χ-Fe5C2。“O 碳化物”包含ε-Fe2C 和ε'-Fe2.2C[35,36]。“O 碳化物”易于在高碳化学势环境中生成,即低温(<200 ℃)和高CO 分压有利于“O 碳化物”生成。与此相反,“TP 碳化物”由于受到动力学因素(晶格变形、碳扩散)的限制,不利于在低温下生成。因此,提高温度时,“O 碳化物”很容易演变成“TP 碳化物”,并在此过程中析出单质C 原子[37]。
已有的实验研究对不同铁碳化物相的作用进行了广泛的探索,结果表明,铁基催化剂在不同的反应条件下由不同的铁碳化物相发挥主要作用,不同的铁碳化物相的作用机制各不相同。在实际反应中,多种铁碳化物相共同作用[38]。在不同还原气氛下,催化剂的反应活性按照χ-Fe5C2、χ-Fe5C2+ε-Fe2C、χ-Fe5C2+Fe7C3的顺序增加[39]。
实验和理论研究表明,ε-Fe2C、χ-Fe5C2和θ-Fe3C 都具有CO 解离和加氢活性,因此,都可作为FTS 反应的活性相,对这三种活性相的研究还没有直接的证据表明单一物相具有某种整体意义上的特征选择性[40]。ε′-Fe2.2C 和ε-Fe2C 可以在较低的温度下形成(200 ℃),具有高度的结构稳定性,是最稳定的铁碳化物相[41]。但同时具有热敏感性,当温度升高到250 ℃时会转化为χ-Fe2.5C 相,因此,ε-Fe2C 可作为低温FTS 催化剂的调控相[42]。作为富碳表面,更倾向于表面碳加氢生成甲烷[43]。χ-Fe5C2具有最低的理论变形能(8.4 × 10-3eV/C)[44],是在H2+CO/CO2混合气氛下最有利生成的物种,由 于H2的存在抑制了χ-Fe5C2向θ-Fe3C 转 变[45]。因此,χ-Fe5C2相也被认为是的FTS 反应的主要活性相[46,47]。其中,(510)面生成CH4具有较高的活化能垒,在热力学上有利于C2+产物的生成[48]。χ-Fe5C2(100)面上优先生成CH4[49]。其中,θ-Fe3C 相是一种亚稳定碳化物铁,它被视为从纯α-Fe 过渡到更富碳的χ-Fe5C2相的中间产物[50]。此外,它还被确定为高温和低碳势下最普遍的相,这表明,θ-Fe3C 可能在高温FTS 反应中发挥重要作用[51],主要生成低碳烯烃[52]。Fe7C3为高温下FTS 反应生成的铁碳化物相,相比于H 辅助CO/CO2解离,Fe7C3表面CO/CO2直接解离能量势垒更低[53]。
1.1.2 FexCy各相间转化
α-Fe 具备优异的CO 解离和加氢性能,已有的实验研究表明,在实际反应中,α-Fe 在催化剂表面存在复杂的动态演化,纯α-Fe 相上CH4的选择性较高[54]。随着反应的进一步进行,α-Fe 晶格中渗碳量增加,α-Fe 向FexCy转化,CHx在FexCy表面链增长生成烃类产物。因此,总结不同Fe/C 下的FexCy表面CO 解离、活化和链增长机理对指导和设计具有优异催化性能的催化剂尤为重要[55-57]。
铁碳化物相形成由许多因素共同决定,如晶粒尺寸、形貌、表面结构和助剂以及其他反应条件(如压力和气体成分)。如图3 所示,改变反应条件,不同铁碳化物相可以互相转化。在低温(<200 ℃)和高碳化学势反应条件下,在热力学和动力学上更倾向于形成八面体碳化物(ε'-Fe2.2C 和ε-Fe2C)。加入助剂和载体可以促进八面体碳化物的形成。在高温和低碳化学势反应条件下,ε'-Fe2.2C 和ε-Fe2C 相会转变为χ-Fe5C2相。随着温度的升高或碳化学势的降低,χ-Fe5C2向θ-Fe3C 转变,这是因为在低碳化学势条件下,θ-Fe3C 比χ-Fe5C2更稳定。Fe7C3是由θ-Fe3C 在高温(>350 ℃)和高碳化学势反应条件下转变而来。越来越多的研究证实,铁碳化物相是FTS 反应中的活性相,在CO 加氢/解离和C-C偶联中起着重要作用,一些研究人员提出,χ-Fe5C2是主要活性相。*C 转化为石墨碳会阻塞活性相表面,进而导致催化剂失活[58]。

图3 金属铁和碳化铁的相互转化示意图Figure 3 Mutual transformation diagram of metallic iron and iron carbide
实验证据表明,活性铁催化剂在FTS 过程中处于动态赝稳态,碳物种在铁碳化物层中缓慢连续地被置换[59],因此,铁碳化物表面的结构稳定性决定了催化剂的稳定性。
1.2 铁基物相表面物质的热化学研究
固体表面的吸附分为物理吸附和化学吸附两大类,物理吸附是多层可逆的,主要作用力是范德华力,物理吸附的吸附速率很快,无需活化能便能很快达到平衡。由于物理吸附不形成新的化学键,因此,不会导致固体表面发生化学重构。化学吸附通常发生在固体表面,且吸附质与吸附剂之间有新的化学键形成,因此,化学吸附通常是单层不可逆吸附。化学吸附需要一定的外部条件,吸附速率较慢[60]。通常用吸附能来衡量化学吸附的稳定性,吸附能越低,吸附越稳定[61]。吸附能受吸附方式,吸附位点,键长,表面覆盖度等因素的影响[62]。通过DFT 计算研究铁碳化物表面物种的吸附主要包括以下三个方面。
1.2.1 CO 和CO2 吸附
CO 在不同铁碳化物的主要稳定吸附表面和主要暴露表面的吸附数据如表1 所示,CO 在Fe(110)表面的吸附较为稳定。Wang 等[63]的研究表明,Fe(110)表面上的C-O 和Fe-C 拉伸频率随覆盖度变化而变化,这是由于在高温下CO 从最稳定的吸附态向不稳定的吸附态的平衡变化引起的。ε-Fe2C 的主要活性面有(011)、(110)、(211)、(121)表面,在(110)、(011)表面的吸附能分别为-2.072、-2.074 eV,最稳定的吸附构型分别对应桥配位吸附和顶配位吸附。CO 在ε-Fe2C(011)面吸附最稳定,暴露面积对总暴露表面积贡献为2%,(101)面为主要暴露面,暴露面积对总暴露表面积贡献达48%。χ-Fe5C2的主要活性面有(510)、(100)、(010)面,主要暴露面有(001)、(110)、(111)、(11-1)面,其中,(510)面是χ-Fe5C2的主要活性面,三配位吸附构型是该表面最稳定CO 吸附构型,吸附能为-2.05 eV,该位点C-O 键得到了很大程度的拉伸,说明该吸附构型有助于C-O 键的解离,进而进行一系列的表面聚合和碳物种加氢反应。(111)面为χ-Fe5C2的最大暴露面,暴露面积对总暴露表面积贡献达36%。

表1 CO 在ε-Fe2C、χ-Fe5C2、θ-Fe3C、Fe3C7 不同表面最稳定吸附位点吸附能、吸附位点键长、振动频率和不同暴露表面面积占总暴露表面面积比Table 1 Adsorption energy,bond length,vibration frequency and the ratio of different exposed surface area to the total exposed surface area of CO on the surfaces of ε-Fe2C,χ-Fe5C2,θ-Fe3C and Fe3C7
θ-Fe3C 主要活性面为(100)、(001)、(010)表面,吸附能分别为-1.77、-1.79、-2.03 eV,最稳定吸附构型为桥配位吸附构型,(111)面为θ-Fe3C 的最大暴露面,暴露面积对总暴露表面积贡献达44%。Fe7C3的主要活性面为(11-1)、(011)、(211)、(1-10)、(101)面,CO 最稳定吸附构型为(1-11)表面的五配位吸附,吸附能为-3.03 eV,与其他碳化物相比,Fe7C3各个表面CO 吸附能力相对较强,不利于生成物的脱附,在(1-11)表面的C-O 键受到了极大的拉伸,因此,该表面更倾向于以CO 直接解离的方式解离。总结各个铁碳化物相吸附规律,从ε-Fe2C 到Fe3C,碳配位数逐渐增加,CO 吸附能也逐渐增加,表面CO 更倾向于吸附在富铁氛围中。由此进一步总结χ-Fe5C2作为FTS 反应主要活性面的原因。(1) C 配位数适中,χ-Fe5C2表面的吸附能力既不过弱也不过强,这有利于促进反应的进行。(2) 形成条件适中,相较于ε-Fe2C,χ-Fe5C2的形成温度为250–400 ℃,该温度氛围下更有利于Fe3O4转变为α-Fe,进而促进χ-Fe5C2相的生成,ε-Fe2C 的形成温度在200–250 ℃。该温度氛围下以Fe3O4为主。因此,不能表现出显著的活性。(3) χ-Fe5C2主要活性表面(510)面为高米勒指数面,较低米勒指数面稳定性较低,更容易产生表面缺陷,有利于CO 的直接解离[76]。后文将重点探究χ-Fe5C2(510)面反应中间物种的吸附和活化。
韩光秀[77]研究了χ-Fe5C2(100)、(110)、(111)、(510)、(111)及(101)晶面对二氧化碳和氢气吸附活化的影响。吸附能计算结果表明,CO2分子和H 原子在(111)和(510)晶面上的吸附最稳定;而且两者在不同晶面上的吸附稳定性与催化剂晶面CO 分子和H 原子转移的电荷量呈正相关。这表明,二氧化碳和氢气在χ-Fe5C2晶面上的吸附是通过催化剂晶面向被吸附分子上转移电荷实现的。CO2在六种晶面上的初始转化路径包括CO2直接解离、加氢生成*HCOO 中间体和加氢生成*COOH中间体三种路径。理论计算结果表明,CO2直接解离比氢辅助解离更容易进行。
1.2.2 H、C、O 的吸附
H2吸附在纯净铁碳化物表面时,首先需要克服H2至H 的微小反应能垒,然后两个氢原子在表面不同吸附位点吸附,单个H 原子与CO2在铁碳化物表面的吸附规律一致,两个氢原子采取单个H 吸附组合的方式进行吸附[78]。
表面或间质C 原子的存在直接影响C-O 键解离机制[79]。主要有两个方面的影响,对于CHx物种的形成,在纯净表面低配位吸附位点上,CO 倾向于以H 辅助的方式解离。当表面存在C 空位,CO 倾向于直接解离成*C 和*O。另一方面,当表面C 空位被占据时,CO 可以以形成乙烯酮(CH2CO)的方式进行表面C-C 偶合。否则更倾向于以CHx(H 辅助)+CHy(表面C)的方式进行表面聚合[80]。
在FTS 反应流程中,O 物种被当作一种希望被除去的物质,原因在于它与希望的目标产物(低碳烯烃等)没有直接关联,并且O 物种在表面的吸附会占据FTS 反应的活性位点,并改变表面电子结构,从而降低催化剂的活性[81]。铁碳化合物表面O 物种可以通过两种方式去除:一是通过与H 结合生成H2O;二是通过与CO 结合生成CO2[82,83]。
O 物种在χ-Fe5C2不同表面吸附的最稳定吸附构型的吸附能和键长等数据如表2 所示,在χ-Fe5C2(510)表面的吸附最稳定,吸附能为-0.99 eV,与CO 在χ-Fe5C2(510)表面相比,*CO 和*O 的最稳定吸附构型都位于三配位位点上,这说明在χ-Fe5C2主要活性表面(510)面上O 物种的吸附会加剧对于表面吸附位点的竞争。对催化剂的性能产生不利影响[84]。

表2 χ-Fe5C2 不同表面O 物种最稳定吸附构型吸附能、键长[84]Table 2 Adsorption energy and bond length of the most stable adsorption configuration of O species on different surfaces of χ-Fe5C2[84](with permission from ACS publications)
1.2.3 CHx的吸附
CH、CH2、CH3是在FTS 甲烷化过程中产生了关键的中间体,Nie 等[85]研究了χ-Fe5C2四种表面上CHx吸附能之间的差异,这些中间体在表面吸附的吸附能如表3 所示。

表3 χ-Fe5C2 不同表面H 和CHx 物种吸附能[85]Table 3 Adsorption energies of H and CHx species on different surfaces of χ-Fe5C2[85](with permission from ACS publications)
结果表明,CHx(x=1–4)的吸附能随着C-H 键数目的增加而逐渐增加。C 和CH 在Fe5C2(510)和(021)表面的吸附能明显小于(001)和(100)表面的吸附能,这可以用吸附位置的差异来解释。C 和CH 在(510)和(021)表面上的吸附位点为4 配位,而在(001)和(100)表面上的吸附位点为3 配位。相比之下,CH4更倾向于吸附在顶配位点。CH4的吸附能明显低于电子不饱和的CHx(x=1–3)。这表明,CH4和χ-Fe5C2表面之间的相互作用非常弱,可以看作是物理吸附,以至于CH4表面的物质很容易从表面脱附。
2 铁基催化剂FTS 反应的密度泛函理论研究
与任何聚合反应一样,FTS 反应包括几个基本步骤:链引发、链增长和链终止[86,87]。中间体的多样性决定了链增长组合的不确定性,这导致FTS 体系是一个庞大的反应网络[88]。加之反应过程中参与反应的活性相可能发生表面重构或相变。用动力学和表征方法进行的实验研究只能提供有限的信息,导致对于FTS 反应机理的研究一直是一个挑战。至今为止已经提出的FTS 反应机理有碳化物机理、CO 插入机理、烷基化机理、烯醇机理、烯烃再吸附机理等[89],其中,被大多数人认可的FTS 反应机理是碳化物机理和CO 插入机理,如表4 所示。

表4 碳化物机理和CO 插入机理对比[86]Table 4 Comparison of carbide mechanism and CO insertion mechanism[86](with permission from ACS publications)
碳化物机理包括CO 完全解离成*C 和*O,*C 和*O 进一步加氢生成CHx和H2O[90-92];氢辅助CO 解离[93]以及H 与非解离CO 的相互作用[94]。如图所示为简化方案。通过FTS 路线合成CHx的不可控表面聚合(图4(a)),使产物分布难以向目标方向进行,产物选择性上限遵循ASF Anderson-Schultz-Flory)分布[95],如下式所示:当α=0.46 时,C2–C4碳氢化合物选择性上限最高(58%)[96](图4(b))。
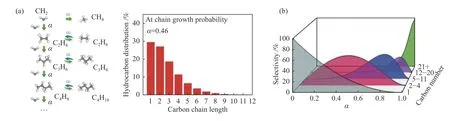
图4 (a)链增长示意图,(b)ASF 分布示意图[95,96]Figure 4 (a) Chain growth diagram,(b) ASF distribution diagram[95,96](with permission from ACS publications)
碳化物的形成机制可以很好地解释长链烃的形成,但不能解释长链烃中含氧化合物的生成[97]。Pichler 等[98]从均相有机金属催化剂作用机理中受到启发,提出了CO 通过直接插入金属-氢键中而引发链增长的机理。与碳化物机理相比,本机理中CO 通过非解离方式插入,更详细地解释了直链产物的形成过程。本机理由于技术及验证方法的局限性,只能根据产生直链烃和支链烃的相对速率来确定支链烃的相对含量,而并未在费托合成条件下获得任何直接的证明。Gaube 等[99]认为,FTS 烃类合成反应的ASF 图是由两种“不相容”机制共同作用的结果。两者都涉及表面烷基化,但在一种情况下,链的生长是通过插入CO 发生的,而在另一种情况下,单体由表面碳烯(CH2)聚合而成。
2.1 链引发
碳化物机理和CO 插入机理都表明,FTS 反应是通过CO 活化引发的。如图5 所示,两种机制的分歧在于,碳化物机制要求CO 首先解离成*C 和*O,而CO 插入机制要求CO 在初始阶段氢化。最近一些基于周期催化剂模型的密度泛函理论研究,通过计算不同反应条件(温度、压力和表面面积暴露比)下所涉及步骤的活化能垒,评估了这些不同活化途径的可能性。在这里,将讨论直接解离路径和H 辅助解离路径的结果。
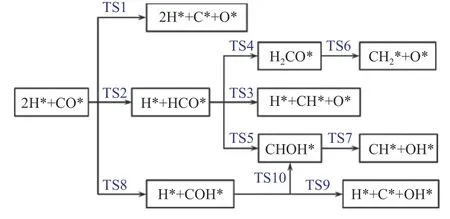
图5 χ-Fe5C2 表面CO 活化机理示意图[100]Figure 5 Mechanism of CO activation on χ-Fe5C2 surface[100](with permission from ACS publications)
2.1.1 CO 通过直接解离机制解离
整理的CO 在四种铁碳化合物不同表面上的最稳定和最活跃位点上的解离、CO 吸附位点、活化能(Ea)、反应能(ΔEr)、CO 解离过渡态的键长(dC-O)数据如表5 所示[101-105],对比数据可以发现,从ε-Fe2C到Fe3C,C 配位逐渐增加,在相应表面最稳定吸附位点上的CO 直接解离的活化能呈逐渐减弱的趋势,表明吸附能越强,CO 直接解离所需要突破的能垒越高。随着CO 在表面吸附配位点的增加,CO 解离能垒减弱,吸附位点特性由稳定吸附转向活跃解离和加氢。

表5 CO 在四种铁碳化合物不同表面上的最稳定和最活跃位点上的解离、CO 吸附位点、活化能(Ea)、反应能(ΔEr)、CO 解离过渡态的键长(dC-O)汇总Table 5 CO dissociation,CO adsorption sites,activation energy (Ea),reaction energy (ΔEr),bond length of CO dissociation transition state (dC-O) at the most stable and active sites of CO on the surfaces of four iron-carbon compounds
Wang 等[65]的研究表明,α-Fe 表面CO 活化机理随表面覆盖度的变化而变化,低覆盖度吸附的CO 分子倾向于在表面解离,高覆盖度吸附的CO 分子倾向于从表面解吸,CO 分子覆盖度介于两者之间时,CO 分子的解吸和解离形成动态平衡。在ε-Fe2C 的四个主要暴露表面上,(011)、(110)和(211)表面CO 直接解离均具有很高的活化能垒,CO 难以通过直接解离的方式进行;(121)表面最稳定吸附构型为四配位位点吸附,具有适中的吸附稳定性和CO 直接解离活性。因此,具有较优异的催化性能。Chen 等[106]的研究也证明ε-Fe2C(121)面具有很好的催化活性。Liu 等[100]对Fe5C2不同表面吸附位点与反应活化能的关系进行了系统的探究,提出了和价键概念,结果表明CO 活化能与和价键呈线性关系。在χ-Fe5C2的九个主要活性面上,(100)和(111)面最活跃反应位点CO 直接解离活化能垒较高,不利于CO 直接解离;(510)面上最活跃反应位点活化能垒较低且对应的最稳定反应位点具有2.57 eV 的活化反应能垒,CO 在表面具有相对稳定的吸附,因此,该表面CO 的解离主要以直接解离方式进行。θ-Fe3C 的几个主要活性面上CO 具有中等的吸附稳定性能和活化能垒,作为铁基催化剂高温活性相具有较高的CO 直接解离活性。Fe7C3表面CO 吸附过强,在最稳定的CO 吸附位点上CO 也具有较高的吸附配位[107]。实验研究表明,CO 在Fe4C 表面直接解离活性很强,相对最低的CO 活化能垒也证明这一论点[108]。
另外,CO2也被用于费托合成制烃类产物的原料气,CO2活化也要经过C-O 键断裂的过程。在*CO2→*CO+*O 步骤中,O 物种既可以持续的吸附在铁碳化合物表面,也可通过进一步加氢生成H2O 而被去除,随后*CO 可通过直接解离或H 辅助方式进一步活化[109]。前人已进行的工作主要集中在χ-Fe5C2相 上,Wang 等[110]研究了在χ-Fe5C2(510)上,最稳定CO2吸附构型分解为吸附的*CO 和*O 的路径。解离后,*O 原子被吸附在4F1 位点,*CO 部分通过稳定在Fe2-Fe4 桥键上的C 原子结合。C 与O1的原子间距离由吸附态的1.34 Å转变为解离态的3.19 Å。在过渡态构型中,C-O1距离为1.76 Å,比CO2吸附结构长0.42 Å,说明在TS 形成过程中C-O 键已经发生断裂。χ-Fe5C2(510)上CO2直接解离的能垒为0.50 eV,反应放热为1.11 eV。
2.1.2 CO 通过H 辅助机制解离
2.1.2.1 CO 在Fe2C 表面通过H 辅助机制解离
Fe2C 在几种铁碳化物相中具有较低的Fe/C 比,因此,在完美的Fe2C 表面上H 更容易直接在表面C上吸附和活化。Cao 等[111]研究了η-Fe2C四个主要暴露表面CO 解离方式,如图6 所示,在完美表面H 辅助CO 解离的活化能垒更低,四个面的*CO →*HCO 路径均具有最低的活化能。其中,(121)表面H 辅助活化能垒最低,CO 直接解离活化能垒为1.85 eV,在2*H+*CO →*H+*CH +*O 步骤具有更低的反应能。还探究了表面存在缺陷时反应路径的变化,如表6 所示,四个缺陷表面CO 直接加氢活化能垒为0.87、1.00、1.03、0.88 eV,较完美表面H 辅助CO 解离活化能垒相比,缺陷表面CO 直接解离活化能垒更低。Chen 等[112]发现,在χ-Fe5C2催化剂表面上,与CO 直接解离有关的铁原子的qB与CO 活化能垒呈线性相关关系。也就是说,铁原子给被吸收的CO 的电子越少,表面铁原子促进CO 解离的能力越强[112]。对η-Fe2C的Bader 电荷(qB)也可以看出,与完美的表面相比,缺陷η-Fe2C 表面所含铁原子电荷更少,同时CO 直接解离能垒更低。含C 空位缺陷表面铁原子促进CO 的直接解离[113]。

表6 η-Fe2C 催化剂完美和缺陷表面CO 活化的活化势垒(Ea)和所涉及表面铁原子的Bader 电荷(qB)[113]Table 6 Activation barrier (Ea) of CO and the Bader charge (qB) of the involved surface Fe atoms on the perfect and defective surfaces of η-Fe2C catalyst[113](with permission from ACS publications)

图6 η-Fe2C(011)、(110)、(211)和(121)表面CO 活化途径的能量[111]Figure 6 Energy diagram of CO activation pathway on η-Fe2C (011),(110),(211) and (121) surfaces Energy zero point is the total energy of free CO and H2 molecules Black line : mechanism I;red line : mechanism II,activate intermediates through *HCO;blue line : Mechanism II,activate intermediates through *COH[111](with permission from ACS publications)
2.1.2.2 CO 在Fe5C2 表面通过H 辅助机制解离
在常见的铁碳化合物中,χ-Fe5C2相的反应温度、压力、Fe/C 比、CO 吸附强度等性能均处于中间状态,这在一定程度上决定了其反应性能,因此,χ-Fe5C2相也被认为是FTS 反应主要活性相。He 等[114]研究对比了χ-Fe5C2相九个主要活性表面最稳定和最活跃位点上通过CO 直接解离机制与H 辅助解离机制解离的不同反应路径及其过渡态能量,对应过渡态如图6 所示,其对应活化能如表7 所示。根据计算的反应能和活化能垒,在不同的表面上发现了不同的活性方式。高指数χ-Fe5C2(510)和相对不稳定的(001)和(221)表面由于存在高活性的CO 吸附位点,对CO 的直接活化更活跃。在χ-Fe5C2(010)、(110)、(11-1)、(-411)和(111)表面具有中等稳定性,这些表面对于直接激活路径来说是相对惰性的,H 辅助CO 解离是可能的。其中,χ-Fe5C2(010)具有较高的H 辅助解离活性。具体来说,CO+H → HCO → CH+O 是χ-Fe5C2(010)、(110)和(11-1)表面上的主要途径;在(-411)表面,CO+H → HCO → CH+O 和CO+H →COH → C+OH 都是可能的路径;而在(111)表面,CO 的活化可能通过CO+2H →HCO+H → HCOH→ CH+OH 的路径进行;此外,在相对稳定的(100)表面上发现CO 活化的可能性较低。Sorescu 的研究表明[115],χ-Fe5C2表面最稳定吸附构型的CO 键解离能也最高。CO 的活化能随着表面原子键总数的增加而显著降低。其中,5F、3F-C 或5F-C 构型的活化能最低。Ozbek 等[116]研究了碳空位对χ-Fe5C2表面CO 吸附的影响,引入碳空位会增加CO 和H 原子的吸附能,有利于CO 直接解离。Chen 等[112]通过Bader 分析和Mulliken 分析研究了CO 活化所涉及的表面铁原子的原子电荷(即给电子容量)的关系,结果表明,两种原子电荷与CO 激活势垒的关系几乎是线性的。参与表面铁原子的电荷主要是给过渡态(TS)提供电子,从而影响过渡态(TS)的稳定性。因此,CO 活化所涉及表面铁原子的原子电荷是描述不同χ-Fe5C2催化剂表面CO 活化的主导因素[117]。

表7 χ-Fe5C2 九个面CO 解离IS(吸附CO 和2H)的吸附能Eads 和TSn 对应的活化能Ea(n)[114]Table 7 Adsorption energy Eads and TSn corresponding activationenergyEa(n) of CO dissociation IS (adsorbed CO and 2H) over nine planes ofχ-Fe5C2[114](with permission from ACS publications)
χ-Fe5C2上CO 解离活性的显著差异和机制的差异说明了控制催化剂形态的重要性。FTS 催化剂暴露面的选择性调整是指导设计催化剂值得关注的方向。
2.1.2.3 CO 在Fe3C 表面通过H 辅助机制解离
目前的工作表明,高温θ-Fe3C 相对CO 键解离具有很高的活性,这是费托反应中必不可少的第一步[118]。一些暴露表面呈现出比α-Fe2C(已知在费托条件下不稳定)和χ-Fe5C2(通常被认为是费托条件下最稳定的碳化铁相)更低的整体CO 解离势垒[119]。Broos 等[120]对θ-Fe3C 武尔夫结构暴露表面CO 解离方式进行了对比研究,研究发现,不同的θ-Fe3C 表面上CO 解离方式不同,如表8所示,在H 辅助的CO 解离在(0-11)、(010)、(001)和(100)表面通过CO → HCO 方式解离是的活化能垒分别为0.62、0.61、1.04、0.75 eV,低于CO 直接解离能垒。在(011)、(110)和(101)表面通过H辅助解离能垒接近CO 的吸附能,表明这些表面是惰性的,CO 活化的可能性较低。

表8 θ-Fe3C 九个表面H 辅助机制活化能和反应能[120]Table 8 Activation and reaction energy of H-assisted mechanisms over nine planes of θ-Fe3C[120](with permission from ACS publications)
2.1.2.4 CO 在Fe7C3 表面通过H 辅助机制解离
h-Fe7C3(211)面的CO 的解离和活化如图7(a)所示,该表面直接解离能垒为1.18 eV,CO → HCO路径活化能垒为0.89 eV,说明该表面更倾向于通H 辅助方式解离,但CO 直接解离反应能更低[121]。在h-Fe7C3(1-11)表面,通过CO → HCO路径活化能垒与直接解离能垒大致相同,沿两种路径活化均倾向于形成CH2物种,如图7(b)所示,对比h-Fe7C3两个表面的CO 解离能量路径可以看出,h-Fe7C3表面具有较为优异的催化反应活性,较强的吸附可能是表面反应难以进行的原因[107]。

图7 (a)h-Fe7C3(211)表面4F2 位点CO 直接或H 辅助解离途径CO 活化机制的能量分布,(b)h-Fe7C3(1-11)表面CO 直接或H 辅助解离途径的构型和能量[107,121]Figure 7 (a) The energy distribution of the CO activation mechanism over the 4F2 site via CO direct or H-assisted dissociation on the surface of h-Fe7C3(211).The configuration and energy of CO direct or H-assisted dissociation on (B) h-Fe7C3(111) surface[107,121](purple : Fe atom ;gray : C atom;red : O atom ;yellow : H atom)(with permission from Molecular Catalysis and ACS publications)
目前,实验研究指导催化剂的设计主要集中在对Fe5C2的性能调控上,因为在常规FTS 条件下Fe5C2相被认为是FTS 反应的主要活性相,理论计算表明,其他铁碳化合物相在特定反应条件下也具有很强的FTS 反应活性。除对单一物相的作用机理进行定性研究外,研究实际反应条件下Fe 基催化剂表面不同物相的分布。以及结合实验探索在反应过程中不同铁碳化合物相表现出的整体催化性能和原因可能是一项必要的工作[122]。
2.2 FexCy 表面低碳化合物的形成
2.2.1 CHx的形成
当碳化物与合成气接触时,首先会发生两种反应。一种是吸附CO 的解离或加氢;另一种是表面C 原子加氢或与CO 耦合[123]。
Song 等[124]研究了CO 在完美η-Fe2C 和缺陷η-Fe2C 表面的活化。发现表面缺陷的存在会对η-Fe2C 表面CO 的活化造成影响,相较于CO 直接解离,在完美的η-Fe2C 表面上,通过HCO*中间体的H 辅助CO 解离是CO 的首选活化途径,但当η-Fe2C 表面存在缺陷是,直接CO 解离是CO 的首选活化途径,因为碳空位的存在导致表面铁原子向吸收的CO 贡献较少的电子。
Pham 等[125]对χ-Fe5C2(510)面碳化物机理CHx形成方式进行了探索,如图8 所示,CH4的形成有三种方式:(1) 表面C 原子在清洁表面上逐步加氢;(2) 游离C 吸附表面C 原子的逐步加氢;(3) 游离C 原子在游离C 吸附表面上的逐步氢化。研究发现,清洁表面上表面C 原子逐步加氢为成CH4的活化能垒最低,为2.39 eV,该表面均表现出较高的CH4形成有效屏障,表明FTS 条件下不利于CH4的形成。

图8 (a)三种方式下甲烷化反应中所涉及的基本步骤的TS 结构,(b)三种情况下CH4 形成的能量分布[125]Figure 8 (a) TS structure of the basic steps involved in the methanation reaction in three ways,Blue : iron atom;gray: C atom;green:C atom involvedinthe reaction;white:Hatom;yellow:Hatominvolvedin the reaction;(b)energy distributionofCH4 formationinthreeways[125](with permission from ACS publications)
Deng 等[126]对Fe3C(100)表面CO 解离路径的研究也表明,相较于CO 直接解离(1.71 eV)和H 辅助解离(1.26 eV)能垒,表面Cs加氢生成表面CsH的活化能垒最低(0.53 eV),同时放热-0.37 eV,是形成表面CH 物种的最有利途径。
对几种铁碳相不同表面CO 活化方式的研究可以看出,(1) 在完美表面,表面C 加氢具有相对较低的活化能垒。(2) 当表面出现碳空位时,CO会在碳空位处优先吸附,此时CO 直接解离是CO的首选活化途径。
为了探究铁碳化合物表面CH4的生成路径和加氢机理,Huo 等[127]通过自旋极化密度泛函理论计算,对Fe2C(011)、Fe5C2(010)、Fe3C(001)和Fe4C(100)上形成CH4的碳路径进行对比研究。首先计算这四个表面CO 解离路径。相比于CO 直接解离(0.95、1.43,1.69、1.36 eV)和H 辅助CO 解离(0.93、1.77、1.13、0.64 eV)。表面C 加氢(0.40、0.31、0.72、0.74)具有更低的活化能垒。与纯净Fe2C(011)、Fe5C2(010)、Fe3C(001)和Fe4C(100)表面相比,C 空位处的CO 吸附具有更大的吸附能(-2.07 vs -1.32、-1.90 vs -1.77 eV、1.92 vs-1.83 eV 和2.20 eV)和更低的解离势垒(分别为0.84 vs 2.79 eV、1.07 vs 1.43 eV、0.91 vs 1.69 eV 和0.93 vs 2.24 eV)。因此,一旦空位位点形成,CO 会先于其他位点吸附解离。由此,空位被重新占据,反应沿② → ③→④ → ⑤ → ⑥ → ⑦→②路径循环进行。
上述发现说明表面C 在催化反应中起着不可替代的作用。在实际复杂的反应进程中,FexCy表面C 可能优先发生加氢反应,参与表面加氢生成CHx物种,CHx形成甲烷或气态烃类脱附后形成表面C 空位。CO 在FexCy表面直接解离或在H2辅助下解离成表面C 物种,表面C 物种在C 空位被重新吸附,由此循环推动反应向下进行[128]。在设计催化剂过程中应根据目标产物的不同设置不同的预处理条件,进而使反应向着热力学有利的方向进行。
2.2.2 CnHy的形成
C2+的形成遵循碳化物机理和CO 插入机理。已有的理论研究建立在这两种机理的假设上。一种是表面Cs(表面C)加氢形成CHx与解离C 形成的CHy相互偶联;另一种是CO 直接与表面C 原子作用形成CsCO,然后再经过直接或H 辅助解离形成CnHy,后续的反应在此基础上循环进行[93,129,130]。
Pham 等[125]对Fe5C2(510)面CnHy的形成进行了理论研究,首先考虑了C 表面生成的CHi(i=0-3)与解离C 生成的CHj(j=0-3)之间基于碳化物机理的10 种C1-C1偶联反应。CHi+CHj化合物之间的C+C 偶联反应的反应势垒最大,生成的C-C 产物非常不稳定。C+CH 和CH+CH 偶联反应相对于其他C1-C1偶联反应具有较低的反应能垒和活化能垒。随后探究了CO 插入的反应活化能。如图9(a)所示:C+CO 偶联反应的反应势垒最大,CH+CO 偶联反应的单个反应势垒和有效势垒相对低于CH2+CO 和CH3+CO 偶联反应。该步骤与C+CH 和CH+CH 偶联反应具有相似的单独反应屏障和有效屏障。在此基础上,进一步研究了基于碳化物或CO 插入机制的CH +CO → CCH+O 反应途径。CO 插入路径的整体势垒为1.56 eV,比碳化物路径高0.47 eV。这表明χ-Fe5C2(510)表面的C1-C1偶联反应机制主要为碳化物机制。先前的研究表明,CO 插入机制是χ-Fe5C2(001)表面C1-C1耦合的主要机制。这说明C1-C1偶联机制对χ-Fe5C2晶面高度敏感。此外,还对该表面与已报道的FTS 催化剂表面的选择性进行了比较和讨论。如图9(b)所示,该表面显示出出乎意料的高C2+选择性。这有力地表明,操纵χ-Fe5C2催化剂的晶面可以有效地调整催化剂的活性和FTS 选择性。

图9 (a)χ-Fe5C2(510)表面CH+CO → CCH+O 形成途径的能量和结构:碳化物机制(红色实线)和CO 插入机制(蓝色虚线)(零点能量也包括在内,蓝色:铁原子;灰色:C 原子;绿色:参与反应的C 原子;白色:H 原子;红色:O 原子)(b)C1-C1 偶联反应的有效势垒(Eeff,C-C)和反应势垒(Ea),以及对χ-Fe5C2(510)和χ-Fe5C2(100)表面的反应物能(Ei+Ej)[125]Figure 9 (a) The energy and structure of CH+CO → CCH+O formation pathway on χ-Fe5C2 (510) surface : carbide mechanism(red solid line) and CO insertion mechanism (blue dotted line).The zero energy is included (Blue: iron atom;gray: C atom;green: C atoms involved in the reaction;white: H atom;red: O atoms)(b) Effective barrier (Eeff,C-C) and the reaction barrier (Ea) for the C1-C1 coupling reaction,as well as the energies of reactant (Ei+Ej) for χ-Fe5C2(510) and χ-Fe5C2(100) surfaces[125](with permission from ACS publications)
为了探究χ-Fe5C2不同表面C1-C1偶联机制对χ-Fe5C2晶面敏感性,Pham 等对比探究了几个χ-Fe5C2表面CHx+CHy耦合的势垒和反应能,如表9 所示,可以看出,在(510)、(021)和(001)表面上,C-CH 和CH-CH 反应是主要的C1-C1耦合途径,因为它们具有最低的活化能垒,而在(100)表面上,CH2-CH2是主要的C1-C1耦合途径。由于主耦合途径的反应速率比其他途径的反应速率大几个数量级,因此,表面的C1-C1耦合速率将近似等于主耦合反应的速率。耦合结果表明,阶梯状(510)和(021)表面的C1-C1耦合反应速率远高于阶梯状(001)和(100)表面的C1-C1耦合反应速率。χ-Fe5C2催化剂的阶梯式表面具有较高的C1-C1偶联活性,导致CH4选择性降低。这意味着较大的催化剂颗粒可能具有较低的CH4选择性。Cao 等[93]对Fe5C2(001)上FTS 合成链生长机理的研究表明,Fe5C2(001)表面除了CH*加氢生成碳氢化合物外,表面O*加氢生成水对产生和维持表面稳定性和反应活性也很重要。在较高的复盖率下,表面O*易加氢生成表面OH*。
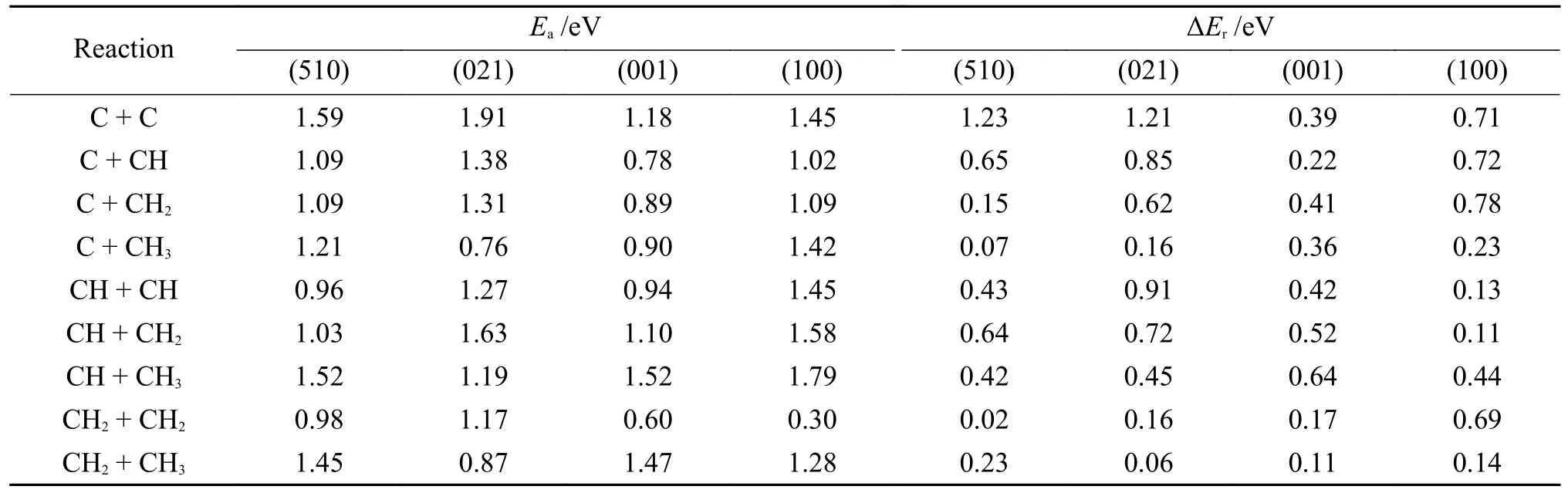
表9 χ-Fe5C2 表面CHx+CHy 耦合的势垒和反应能[131]Table 9 Barrier and reaction energy of CHx+CHy coupling on χ-Fe5C2 planes[131](with permission from ACS publications)
在实际复杂反应中,不同表面对某一产物的动力学贡献缺乏定性分析。为了探究这一问题,Kleis 等[132]对χ-Fe5C2不同表面对CH4和C2+产物的贡献率做了动力学分析,如图10 所示。结果表明,(111)表面是Fe5C2粒子暴露强度最大的表面(24.0%)。它在C2+产物的生成物中占53.3%,在CH4的生成中占1.8%。值得注意的是,(10-1)表面轻微暴露在Fe5C2颗粒上(3.2%),然而,它们在C2+产物形成却有着46.5%的贡献率。表明(10-1)表在Fe5C2颗粒的宏观动力学中起着重要作用,这反映在(110)、(010)和(11-1)表面对Fe5C2颗粒生成CH4的动力学贡献最大(图10(a));而表面(10-1)和(111)在C2物种形成的动力学中占主导地位(图10(b))。其余已探索的表面在生成CH4和C2物种时不具有动力学竞争性。
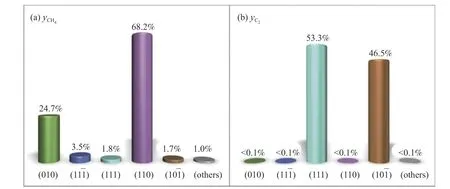
图10 在pH2/pCO2 条件下,各表面对Fe5C2 颗粒的动力学贡献(a)CH4 组和(b)C2+ 组[131]Figure 10 Kinetic contribution of each plane of Fe5C2 particles under=pH2/pCO2 conditions (a) CH4 and (b) C2+pCO=1.0 * 10-4 Pa and T=550 K[131](with permission from ACS publications)
关于Fe5C2表面C-C 偶联机理前人已经做了相对充分的研究,但对其他铁碳化合物C-C 偶联机理的研究还相对缺乏,Deng 等[126]研究了Fe3C(100)表明CnHy形成机理,结果表明,CO 吸附产生的表面乙烯基CsCO 是C2的第一个表面物质,是生成C2Hx的重要中间体。初始表面C2Hx由CsCO 氢化反应形成,表面CsCO 作为C2Hx生成的重要中间体,既可解离也可氢化。CsCO 直接表面解离生成CsC 和O 的势垒为1.60 eV,而CsCO加氢生成CsCHO 的势垒最低,为0.40 eV,放热为0.11 eV。从含氧CsCHO 中间体开始,值得注意的是,表面C2Hx物种,CsCH,CsHCH 和CsH2CH,既可以来自氢化解离途径,也可以来自解离氢化途径。根据可比较的有效势垒和反应能,C2Hx的形成取决于CO/H2比。为了探究CO/H2比和压力等因素对碳碳偶联活性造成的影响。Yang 等[133]比较了Fe3C(010)不同Fe/C 表面C-C 偶联机理,结果表明,Fe/C 端Fe3C(010)-0.25表面形成1-丙基的总势垒为0.51 eV (CO+3H+2Cs),低于Fe3C(010)-0.00表面形成CH2CH 的势垒(1.34 eV)。富碳Fe3C(010)-0.25表面比富铁的Fe3C(010)-0.00表面更有效地进行链增长。因此,在实验中应采用合理的H2/CO 比值,以增加氢吸附和甲基生成的可能性。
2.3 链终止
到目前的研究还没有涉及到链终止路径的理论工作,很大一个原因是FTS 机理的探索工作还不够系统[134],在目前的FTS 机理中,碳化物机制可以很好地解释碳氢化合物的形成,但不能解释氧化物的形成。CO 插入的情况正好相反。但至今还没有一种机理能够阐明所有的产物[135]。因此,需要一种结合碳化物路径和CO 插入路径优点的统一新机制,在此基础上再对链终止方式进行系统探索。
3 铁基催化剂性能调控机制
3.1 催化剂颗粒调控
3.1.1 尺寸效应
Kleis 等[136]通过计算一氧化碳和氧气在一系列金纳米颗粒上的吸附能量,计算了从小团簇到固体化学键合中的有限尺寸效应。计算结果得到固体与气相的边界为2.7 nm,在此尺寸之上,两个探针吸附剂,O 和CO 的表面化学性质与扩展表面难以区分。在此阈值以下,存在强烈的有限尺寸效应,我这可能与相对于簇的大小的吸附事件的电荷密度响应的空间范围有关。碳化铁团簇的尺寸效应表现在几个方面:(1) 随着团簇变大,平均Fe-Fe 键长继续增加,并在团簇大小达到40 个原子左右时收敛;(2) 铁原子的磁矩随原子数的增加总体呈减小趋势,但波动较大;(3) 随着团簇尺寸的增加,凝聚能(CE)降低(量级增加),这意味着更大的团簇具有更高的解离能。
以往的实验工作表明,FTS 活性和选择性与铁基催化剂的粒径密切相关[137]。Mabaso 等[138]的研究表明,粒径小于7–9 nm 的铁纳米颗粒催化剂比含有较大颗粒的催化剂具有更低的转换频率(Turnover Frequency,TOF)和更高的CH4选择性。烯烃选择性不受粒径的影响,而较小的铁纳米颗粒具有较低的链生长概率和较高的CH4选择性。进一步的研究表明,当铁粒径从2.4 nm 增加到6.2 nm 时,TOF 增大,然后在粒径为11.5 nm 时几乎保持不变。CHx覆盖度随粒径增大而减小,这是由于较小的颗粒具有较高数量的高活性低配位。研究还证实,随着CHx的覆盖,翻转频率增加。颗粒越大,H 覆盖率越低[139]。Bader 电荷分析表明,Fe 向C 转移的电子与团簇大小成正比。由于铁原子的净电荷与其上的反应能垒密切相关[140],因此,控制碳化铁团簇的大小是调节其催化活性的有效方法。
3.1.2 形貌调控
Zhao 等[141]研究了Hägg 碳化铁(χ-Fe5C2)的表面形貌的影响因素,研究结果表明,CO 和H2/CO气体压力对碳化学势(uc)的影响远大于温度,较低的温度和/或较低的总压会导致较高的碳化学势和较高的富C 面稳定性。CO 中加入少量H2(H2/CO=1∶10)可导致碳化学势(uc)大幅度降低。当H2/CO比值在600 K 和1 atm 时 从1∶10 增加到10∶1 时,碳化学势(uc)先缓慢增加,然后在H2/CO=2.5 时达到最大值后急剧下降。当H2分压非常高(H2/CO=5∶1)时,χ-Fe5C2相热力学不稳定。在CO 和H2/CO预处理条件下,χ-Fe5C2相具有相似的形貌,即只有(010)、(011)、(100)、(111)、(001)、(133)、(113)和(113)可以以Wulff 形状暴露(如图11 所示),但在不同Fe/C 比下,各表面的暴露比例有所不同。CO 预处理有利于富C 端,而合成气预处理有利于贫C 端。表面碳的高加氢活性可能解释了CO 预处理比合成气预处理具有更高的FTS 初始活性[142]。

图11 (a)CO 和合成气预处理下Hägg 碳化物的形貌(括号中给出的指标表示对应的米勒指数,指数的第二项提供了对应的表面a=Fe/C 比,指数的第三项提供了每个暴露表面对总表面积的贡献);(b)图(a)中构造的8 个Wulff 暴露表面对应碳化学势(μc)最稳定端的表面结构(图左括号中给出的指标表示每个表面的米勒指数,每个结构下的数字分别为对应表面a=Fe/C 比,蓝球表示Fe 原子,黑球表示C 原子)[142]Figure 11 (a) Morphologies of Hägg carbide under CO and syngas pretreatments (indices given in parentheses indicates the corresponding Miller index.The second number is the corresponding surface Fe/C ratio.The third number is the contribution of each plane to the total surface area).(B) The most stable terminations of the structures from Wulff construction in Figure (a) at corresponding carbon chemical potential(mc) (indices on the left of the figure indicates the Miller index of each plane.The number under each structure is the corresponding surface Fe/C ratio,blueballs for Fe atoms and black balls for C atoms)[142](with permission from ACS publications)
3.2 金属助剂调控
在催化剂中添加助剂通常是为了优化FTS 的产物选择性和催化活性。助剂可作为结构或电子改性剂、稳定剂或抗毒催化剂用于FTS。一般来说,助剂分为三类:影响金属载体相互作用的结构助剂、过渡金属助剂、影响催化剂表面电子密度的电子助剂[143]。
3.2.1 结构助剂
结构助剂指通过改变催化剂的化学组成、化学或晶格结构、孔结构、分散状态、机械强度等并提高活性组分的分散性和热稳定性的助催化剂[144]。通常包括金属氧化物和非金属氧化物载体(如C 和Si 载体等)[145]。结构助剂可以改变催化剂的吸附和反应活性,其吸附性作用规律可以用火山曲线描述,如图12 所示,如果催化剂与吸附质之间的相互作用过强,这有利于反应物分子的吸附和活化,但产物的较难脱附也会影响整体的催化性能。相反,如果催化剂与吸附质之间的相互作用过弱,将利于产物在催化剂表面的脱附,但较高的反应能垒同样不利于整体催化性能的提升[146]。

图12 催化剂活性与表面结合强度的关系[146]Figure 12 Relationship between catalyst activity and surface bonding strength[146](with permission from ACS publications)
在铁基催化剂中加入SiO2对FTS 性能的影响也得到了广泛的研究。通常,SiO2掺入到铁基FTS 催化剂中会导致FTS 活性降低。Bukur 等报道,随着SiO2含量的增加,FTS 活性和水煤气变换(Water Gas Shift,WGS)反应活性均降低。此外,随着SiO2含量的增加,烯烃加氢和异构化的二次反应得到加强,催化剂的还原受到抑制。这与Cu-Fe 催化剂表现出的催化性相似[109,147]。
3.2.2 过渡金属助剂
Cu 是常用的FTS 过渡金属助剂,Cu 作为RWGS活性相[147],可在较低温度下促进氧化铁的还原,进而促进FTS 活性相的形成[148]。虽然铜在促进催化剂还原方面的作用已被广泛接受,在铁基催化剂中加入Cu 会导致烃产物的平均分子量增加[149];Tian 等[150,151]的研究表明不同的Fe/C 表面Cu 与表面原子作用的相互关系不同,这种电荷转移的不同行为来自于Fe、C 和Cu 原子之间电负性的差异。Fe-Cu 作用的Cu-Zn 协同作用通过提高了催化剂的加氢能力,因此,在Cu 促进的Fe-Zn 催化剂上有很高的CH4选择性[152]。
Zn 促进剂对χ-Fe5C2的稳定作用通过多种催化剂表征和DFT 计算得到了证实。可以得出以下结论:Zn 助剂的加入增强了CH 中间体的加氢能力,促进了反应的正向进行,阻止了表面碳的沉积[153]。锌改性降低了表面吸附氧原子的氢化障碍,使表面H2O 分子失稳,共同降低了碳化铁氧化的可能性,同时减小了χ-Fe5C2表面氧原子的覆盖。这有利于反应过程中其他中间体的吸附,从而促进了催化剂的长期稳定性[154]。
Al 常被用来调控催化剂表面碱度[155],Fe-Al2O3之间具有强烈的相互作用,Al2O3的加入抑制了FeO 还原为Fe,阻碍了铁催化剂的渗碳。此外,Fe-Al2O3的强相互作用减弱了催化剂的表面碱度,促进了H2的吸附,抑制了CO 的吸附,使得FTS和WGS 活性降低。而Fe-Al2O3的强相互作用抑制了铁碳化物的再氧化,稳定了FTS 活性位点,提高了催化剂的稳定性。Al2O3可以促进H 在铁基催化剂表面的吸附,因此,Al2O3催化剂对轻烃的选择性较高,对重烃的选择性较低[156]。
在FTS 中碳载体能有效促进碳化物物种的形成[157-159]。特别是介孔碳,由于其大比表面积和孔体积,高孔隙率,促进气体和液体扩散和物质通量的良好可达性,已被证明具有优势[160-162]。此外,碳的梯度孔隙率不断降低对液体或气体的质量传输阻力,以及催化反应中催化剂利用率和功率密度的增加,使碳与其他载体相比具有优势[35,100,102,160,161]。
第八族贵金属Rh、Ru、Pd 和Pt 等常被用作费托催化剂的助剂被广泛研究。根据Fe/M (M 代表贵金属)的比值,催化剂制备方法被分为两类,当Fe 的比例较低时,通过在Si 负载的贵金属催化剂中加入低比例的Fe,制备富贵金属的双金属催化剂;当Fe 的含量占主要时,通过向Fe 中加入少量贵金属,制备富Fe 金属双金属催化剂[162]。
与K/Fe 催化剂类似,Pt/Fe 催化剂可以将产物选择性转移到高分子量烃类。Wang 等[163]的研究发现Pt 的加入有助于Fe3O4的还原,并促进Fe5C2在预处理过程中的分散。此外,Pt 的加入提高了FTS 反应的活性,抑制WGS 反应和降低了甲烷选择性。Pt 促进剂有利于氧化铁的还原和渗碳。总的来说,Pt 能提高铁基FTS 催化剂FTS 活性,降低CH4选择性和烯烃选择性[164]。
Pd 的作用与Pt 类似,都能提高催化剂活性,使产物选择性向长链烷烃转移[165]。Ru 有助于Fe3O4的还原,这有助于FTS 活性相的形成[166]。Rh、Ru、Pd 和Pt 助剂都对烯烃/石蜡比有负面影响。所有助剂都显著提高了CO 的氢化速率,这是由于贵金属助剂的加入增加活性金属中心的数量,进而促进了CO 的还原和CO 的分散[167]。
3.2.3 电子助剂
电子助剂被用于提高催化剂对烯烃的活性和选择性已被广泛研究。使用最多的是元素周期表第一组的碱金属,碱金属被视为金属中心的电子供体,有助于增强CO2等酸性气体的吸附和抑制H2和烯烃的吸附,提高催化剂表面碳氢比从而导致更高的烯烃含量[112]。钠和钾的存在提高了Fe-C 键的强度,与加氢屏障相比,碱金属促进的催化剂导致较低的吸附能,有利于烯烃脱附,且不需要的进一步加氢[70]。
其中,钾(K)被认为与该组其他碱金属相比具有最佳的成本效益比。通过对Fe(110)[121]、Fe(100)[123]和Fe(111)[130]的单晶研究和使用多晶铁[96]进行的实验已经证实钾增强了CO 吸附强度的及其分解速率,显然,钾增加了CO 的吸附,从而减少了氢的相对吸附量。从这个角度看,钾降低了仲烯烃的氢化反应。如图13 所示,钾将电子捐赠给铁,这将促进CO 的解离,CO 倾向于接受来自它的电子。例如,给电子的Na 和K 使铁的亲电性降低,导致H2亲和力降低,催化剂表面的H 浓度降低,这有利于通过抑制氢化来生产烯烃。Zhao 等[168-170]的研究表明,K2O 助剂降低了CHxOH 的CO 键解离障碍(x=0–2),使其首选路径转变为HCO →CHOH → CH+OH 和HCO → CH2O → CH2OH →CH2+OH。这促进了表面C1 物种的形成;它可以用来引发碳链生长的碳碳偶联反应。这与实验观察到的钾催化氧化产物选择性降低与非催化氧化产物选择性降低相一致。K2O 吸附对Fe5C2近表面碳原子和吸附表面氢原子的影响强于远表面碳原子。Zhao 等[171]的研究表明,钾在随后的铁氧化物渗碳和还原过程中促进了新的活性位点的形成,促进了形核位点的快速形成,生成铁碳化物的小晶体。另一方面,K 助剂的加入会增强O 物种在表面的吸附,O 吸附原子上的电子密度增加,氧物种的吸附会降低CO 在催化剂表面的覆盖度。因此,探究K/Fe 催化剂表面氧物种的去除是决定该催化体系的一个不可忽视的问题。Na 或K 助剂的另一个功能是促进生成χ-Fe5C2相,这是FTS的活性相,归因于铁催化剂还原性的改善[172]。碱金属的负载量也会影响铁基催化剂的活性和选择性。

图13 (左)K 在χ-Fe5C2(100)0.00 上吸附后的电荷密度差图,显示以e/A3 为单位的增加(橙色,红色)和减少(蓝色)区域;(a)中的采样平面穿过山谷并经过K,(b)中的采样平面穿过山谷并经过K;(右)预覆盖K 的χ-Fe5C2(100)表面上O 吸附电荷密度变化的(a)侧视图和(b)俯视图,红色(绿色)等值面表示增加(减少)0.015 e/A3;(c)χ-Fe5C2(100)上吸附K 和吸附O 相互作用时电荷密度变化的侧视图和(d)俯视图,红色(绿色)等值面表示增加(减少)0.005 e/A3,Fe 和C 原子分别为紫色和橙色,K 和O 的位置被标记[122]Figure 13 (left) The charge density difference of K adsorbed on χ-Fe5C2 (100)0.00 shown the increase (orange,red) or decrease (blue)regions in e/A3.The sampling plane in (a) crosses the valley and through K.The sampling plane in (b) crosses the valley and through K.(right) (a) side view and (b) top view of the change of O adsorption charge density on χ-Fe5C2(100) surface pre-covered with K.The red(green) isosurface indicates an increase (or decrease) of 0.015 e/A3.(c) The side view and (d) top view of the change of charge density in the interaction between adsorbed K and adsorbed O on χ-Fe5C2(100).The red (green) isosurface indicates an increase (decrease) of 0.005 e/A3.The Fe and C atoms are purple and orange,respectively.The positions of K and O are marked[122](with permission from ACS publications)
4 总结与展望
铁基催化剂是目前在中国广泛使用的FTS 催化剂,如何开发设计具有优异催化活性的铁基催化剂具有广阔的研究前景。因此,对铁基催化剂作用机理的研究显得尤为重要。在这个问题上,理论计算建立了微观尺度与宏观尺度之间的桥梁。
铁基催化剂在反应过程中存在复杂的物相动态演变,不同物相起到的作用不同。不同物相之间可以协同作用,最终表现出催化剂的整体催化性能。已有的研究表明,通过对铁碳化合物相的调控,能明显改善Fe 基催化剂的催化性能。实验观察到稳定存在的铁碳化合物相主要有Fe2C、Fe5C2、Fe3C、Fe7C3。对不同相态FTS 反应能量路径的研究表明,将Fe5C2作为FTS 的主要活性相的说法需要添加一定的限制条件,原因在于不同铁碳化合物相的形成和发挥独特的催化性能需要的反应条件不同。
Fe2C 具有优异的FTS 反应活性,CO 在该表面主要以直接解离方式活化。Fe2C 是最先形成的铁碳化合物相,但其生成温度范围较窄(200–250 ℃)。当温度高于250 ℃时,Fe2C 转化为Fe5C2。且Fe2C 生成温度与RWGS 反应温度重合。在实际反应中可能被Fe4O3包覆而无法发挥显著的催化作用。Fe5C2是FTS 反应条件下稳定存在的相,Fe5C2具有显著催化活性的原因有两点:(1) FTS 适宜反应温度与Fe5C2形成温度重合,Fe5C2在实际反应中成为主要铁碳化合物相;(2) Fe5C2吸附能力适中,不同表面可以以不同方式解离和活化。因此,表现出较好的整体催化活性。Fe3C 作为高温条件下的FTS 活性相,表面吸附能力较强整体,CO 活化能垒较低,在反应条件下控制形成更多的Fe3C物相可能对整体反应有利。
在对CHx加氢和对C-C 耦合机理的研究中,表面C 发挥着重要的作用。CO2在铁碳化合物表面活化过程中,铁碳化合物表面的C 可以优先加氢,并暴露出C 空位。随后CO2在C 空位处吸附,CO2进一步活化以维持铁碳化合物表面形貌,通过表面C 循环推动CO2加氢反应循环进行。碳物种在铁表面的吸附较强,在钴表面的吸附适中,在C 表面的吸附极弱。对于链增长反应而言,从各物相的研究结果来看,表面结构对C-C 偶合反应的影响显著,即使同一种物相,不同晶面的反应性质相差巨大,寻找控制链增长的关键结构因子是催化剂优化的关键之一。最后总结了结构助剂、电子助剂、过渡金属助剂对铁基催化剂的影响,结构助剂主要起到负载活性中心的作用,通过改变催化剂的比表面积、表面碱度、吸附能力等改善催化剂的催化性能。电子助剂通常指第一组的碱金属,通过改变催化剂电子转移能力来改善催化剂的催化性能;过渡金属催化剂则通过改变活性中心的性质来改善催化剂的催化性能。目前对于各种费托助剂的理论计算研究报道相对较少,催化剂的理性设计需要对金属助剂的作用机制进行全面深入的认识。
虽然研究者运用DFT 计算探究铁基催化剂的相关反应已经取得了一系列进展,但由于碳扩散动力学和复杂的表面重构、表面吸附物种和晶格缺陷的干扰以及计算方法的限制,真实条件下出现的碳化铁催化剂的准确原子级结构仍然不清楚,它们的形成复杂的机制,需要进一步研究。关于铁基催化剂碳作用机制需要解决的问题主要有:C 在α-Fe 团簇表面的渗透;C 在α-Fe 团簇内部的扩散;C 引起的FeCx表面重构;碳势与FeCx物相转变的关系;C 对铁基催化剂电子性质的影响。虽然在理论上可以通过合理的简化来评估碳化铁表面的CO 活化和CH4选择性,但FTS 对碳化铁的详细机制尚不清楚。因此,利用理论方法建立FTS 中铁碳化物的结构-性能关系仍然是一个相当大的挑战。另外,表面过度碳沉积导致的催化剂失活也是催化剂能否大规模应用的一个决定性因素。要探究积碳机理,需要对FTS 反应活性相不同表面FeCx的形成进行系统的动力学和热力学分析。并通过控制表面C 覆盖度探究反应催化剂的活性和选择性,进而采取针对性的方法避免催化剂的失活。
在实际的反应中,很难控制单一活性相,但是由于各物相不同的晶体结构和电子性质,其催化作用具有差异性,表面科学模型催化研究可能是研究这一问题的方法之一,对于理论计算研究而言,探索不同铁基物相的结构,尤其是表面结构与催化性能之间的关系能有效帮助研究者建立合理的构效关系,从而为催化剂结构的调控提供一定的理论基础。然而在具体研究过程中,复杂的表面结构成为该体系理性认知的难点,结合体相信息,从复杂的表面配位环境中抽提出具有物理意义的普适性规律将对铁基费托催化的研究和认识提供很大的助益。
