核壳型MOFs@离子液体复合材料的制备及其常压下催化CO2 环加成反应性能探究
2023-11-21胡凤群邱明月李向远李剑川史利娟段小川
胡凤群,邱明月,易 群,3,张 鼎,李向远,李剑川,史利娟,,*,段小川,*
(1.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西太原 030024;2.武汉工程大学 化工与制药学院,湖北 武汉 430205;3.山西浙大新材料与化工研究院,山西太原 030000;4.天脊煤化工集团有限公司,山西 长治 046000)
二氧化碳(CO2)是一种主要的温室气体,同时也是一种无毒、经济且丰富的C1 资源,可以转化为许多高附加值化学品[1]。其中,CO2与环氧化物通过环加成反应生成环状碳酸酯是一种原子经济性100%的绿色工艺,环状碳酸酯在工业上有着广泛的应用,可作为锂离子电池、金属提取、化妆品、精细化学中间体和无质子溶剂的电解液等[2]。然而,CO2固有的热力学稳定性和动力学惰性使其难以被活化,这给固定CO2带来了巨大挑战[3]。为了克服这一难题,已经开发了大量的均相和非均相催化剂来促进CO2环加成反应。鉴于均相催化剂存在产品分离、纯化困难等问题[4],用于CO2环加成反应的多相催化剂逐渐受到更多关注,如金属有机框架材料(MOFs)、共价有机框架材料(COFs)、负载型离子液体等材料[5]。然而,当前开发的非均相催化剂大多具有种类单一的活性组分(如Lewis 酸或Lewis 碱),导致催化剂催化效率相对较低,在催化过程中需要苛刻的反应条件(例如高温、高压等)或者添加助催化剂[6]。因此,将Lewis 酸性中心和碱性亲核官能团结合,构建Lewis 酸碱位点高效协同的双功能材料,是开发高效、有前景环加成反应催化剂的有效策略[7]。
MOFs 因为具有较大的比表面积、可调的孔结构以及固有的Lewis 酸性金属中心,成为一类典型的Lewis 酸性非均相催化剂[8]。离子液体(ILs),具有亲电阳离子和Lewis 碱性卤素阴离子,在CO2/环氧化物环加成反应中表现出良好的催化活性[9]。将离子液体和MOFs 材料有效结合,有望构建得到Lewis 酸碱位点双功能的非均相催化剂[10,11]。当前,主要是通过共价接枝将ILs 修饰于MOFs 配体[12,13],或通过浸渍法将ILs 直接引入到MOFs 孔道中[14]。然而,上述两种方法常导致明显的比表面积下降和孔道堵塞,影响反应底物和产物在复合材料中扩散,抑或造成离子液体流失,从而降低Lewis 酸碱协同效率,导致催化效率仍不理想[6]。
核-壳型复合材料,可以有效保持内外材料的多孔结构,有效解决传统复合材料比表面积下降和孔隙堵塞等问题[15,16]。例如Tsal 等[17]通过涂层的方法合成了SiO2@ZIF-67 核壳微球,SiO2@ZIF-67在涂层处理后仍保留了ZIF-67 的孔隙率和比表面积。当前报道的核壳型MOFs@ILs 复合材料大多数是通过浸渍法将离子液体单体直接负载于MOFs表面,例如Zhao 等[18]通过浸渍法将离子液体组装与MOF-808 表面,制备了核壳结构的MOFs@ILs复合材料用于CO2吸附。然而通过浸渍法制备的MOFs@ILs 复合材料存在离子液体易流失、在反应条件下不稳定的缺点,无法用于CO2催化转化。基于此,实验首先将离子液体和对苯二甲醛共价组装生成聚合物,然后将聚合物包覆于MOFs材料表面,有效避免了 ILs 流失。此外,材料在保留了MOFs 的晶体多孔结构和高比表面积的同时,从 ILs 结构中引入亲核位点,通过Lewis 酸性位点和亲核位点的协同作用有效提高环加成反应效率。
1 实验部分
1.1 实验试剂
对苯二甲醛(99%),购于百灵威科技有限公司;九水合硝酸铬(99%),无水乙醇、无水甲醇、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)均为分析纯,购于国药集团化学试剂有限公司;3-氯丙胺盐酸盐、氢氧化钠均为优级纯,购于Adamas 试剂有限公司;乙腈,分析纯,购于天津大茂化学试剂有限公司;氮气(N2)、二氧化碳(CO2),≥99.999%,购于太原钢铁公司。
1.2 ILs/MOFs 复合材料的制备
1.2.1 DAIL 的合成
1,4-丁基二咪唑的合成(图1)[19]:称取咪唑(13.616 g,0.2 mol)、氢氧化钠(9.6 g,0.24 mol)、二甲基亚砜40 mL 加入到150 mL 圆底烧瓶中,控制反应温度为60 ℃,磁力搅拌4 h。然后将1,4-二氯丁烷(10.161 g,0.08 mol)加入到恒压滴液漏斗中,缓慢滴入混合物中,继续反应6 h。反应结束,冷却至室温,将反应混合物缓慢的倒入冷水中并使用玻璃棒搅拌,出现白色固体,抽滤并用冷水洗涤三次,真空干燥24 h。得到白色固体产品即为1,4-丁基二咪唑。1H NMR (400 MHz,DMSO-d6)):δ7.58 (s,1H),7.12 (s,1H),6.85 (s,1H),3.93 (s,2H),1.59 (s,2H)。

图1 1.4-丁基二咪唑反应方程式Figure 1 Reaction formula for the synthesis of 1,4-bis (imidazol-1-yl) butane
DAIL 的合成(图2)[20]:将乙腈(25 mL)、3-氯丙胺盐酸盐(0.06 mol,7.801 g)、1,4-丁基二咪唑(0.02 mol,3.805 g)加入到50 mL 圆底烧瓶中。在75 ℃下氮气保护搅拌48 h。反应结束后,倒掉上层溶剂,下层黄色黏稠物质用乙腈洗涤。洗涤后用少量水溶解,然后加入氢氧化钠水溶液调节溶液pH 值到10。旋蒸,放入真空烘箱中100 ℃干燥12 h。干燥后用无水甲醇洗涤,离心,上清液旋蒸,重复操作三次,直至没有白色NaCl 沉淀产生,将得到的黏稠液体80 ℃真空干燥24 h,得到黄色黏稠状液体,即为双-(3-氨基丙基-1-咪唑)亚丁基二氯盐,简称DAIL。1H NMR (400 MHz,DMSO-d6):δ9.68 (s,1H),7.94 (d,J=8.1 Hz,2H),4.32 (d,J=21.2 Hz,4H),2.63 (s,2H),2.04 (s,2H),1.82 (s,2H)。

图2 离子液体DAIL 的反应方程式Figure 2 Reaction formula for the synthesis of inion liquid DAIL
1.2.2 DAIL/MOFs 复合材料(MIL-101-DAIL)的制备
称取DAIL(0.2 mmol,0.0755 g)溶于2 mL 无水甲醇中,取活化过后MIL-101(Cr)(0.1 g)分散至溶液中,其中,MIL-101(Cr)采用水热法制备[21]。室温搅拌24 h 后将溶液离心,保存上清液,固体用无水甲醇洗涤三次后放入保存的上清液当中,继续搅拌24 h,离心,固体再次用无水甲醇洗涤三次,真空烘箱70 ℃干燥12 h。命名为MIL-101-DAIL。
1.2.3 DP/MOFs 复合材料(MIL-101@DP)的制备
聚合物DP 的制备:将DAIL(0.2 mmol,0.0755 g)、对苯二甲醛(TPA)(0.2 mmol,0.0268 g)溶于2 mL无水甲醇中,放入40 ℃恒温培养箱中,反应48 h。命名为DP-1。将DAIL 和TPA 浓度增大为0.15 mol/L,命名为DP-2。反应方程式如图3 所示。

图3 聚合物DP 反应方程式Figure 3 Reaction formula for the synthesis of polymer DP
MIL-101@DP-X 的制备:将活化过后的MIL-101(Cr)(0.1 g)分别加入到上述DP-X 的溶液当中,室温下搅拌24 h,离心,保存上清液,固体用无水甲醇洗涤三次后放入保存的上清液当中,继续搅拌24 h,离心后固体再次用无水甲醇洗涤三次,真空烘箱70 ℃干燥12 h。得到MIL-101@DP-X。
1.3 催化剂的表征
样品的结构分析使用Bruker Vertex 70 型傅里叶变换红外光谱仪,测试之前样品在真空条件下70 ℃干燥12 h,溴化钾和材料按质量比100∶1 的比例混合压片,压片后在红外灯下烘干,扫描400–4000 cm-1。
样品的核磁共振波谱分析使用德国Bruker Avance III 型仪器。
样品的晶体结构分析使用Rigaku 公司UltimaⅣ 型X 射线衍射仪,测试之前样品在真空条件下70 ℃干燥12 h,扫描速率4(°)/min,2°–20°扫描。
样品的形貌使用蔡司Sigma 300 型场发射扫描电镜进行观察。
样品的比表面积和孔容分析使用北京精微高博科学技术有限公司的JW-BK200 型全自动物理吸附仪,测试之前在120 ℃、10 h 条件下进行活化处理。比表面积计算采用BET 方程,孔径分布计算使用非密度泛函理论(NLDFT)法。
样品的热稳定性分析使用TA SDT650 型热重分析仪,测试气氛为N2,升温速率10 ℃/min,温度30–800 ℃。
1.4 催化性能评价
以环氧氯丙烷(ECH)为底物,在无助催化剂条件下考察了该催化剂催化CO2环加成反应性能,环加成反应方程式如图4 所示[22]。
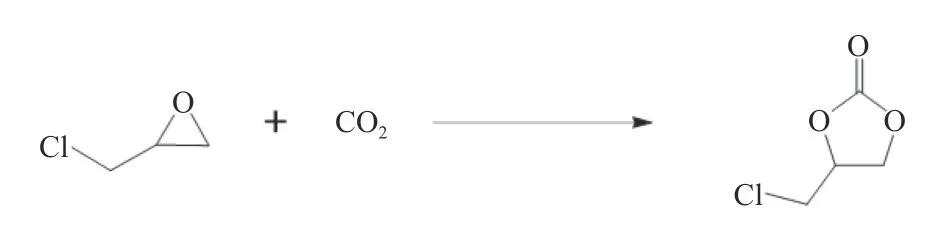
图4 ECH 和CO2 环加成反应Figure 4 The cycloaddition of ECH and CO2
反应在10 mL 史莱克管中进行,在无溶剂的情况下,将ECH(10 mmol,0.925 g)和催化剂(10 mg)加入有磁子的史莱克管中,装上乳胶气球和磨口玻璃塞,用封口膜密封,抽真空并充入CO2,重复三次以除去反应装置中的空气,放入油浴锅中。反应结束后在冰水浴中冷却,释放未反应的CO2。将催化剂通过离心从悬浊液中分离出来,经过无水甲醇洗涤和真空干燥后重复使用。上清液采用CDCl3做溶剂,并用1H NMR 测定反应的转化率和选择性。产物核磁数据:1H NMR (400 MHz,CDCl3):δ5.05 (m,1H),4.65 (dd,1H),4.43 (dd,1H),3.89 (m,2H)。
转化率(%)、选择性(%)分别按式(1)、(2)计算[23],其中,C0和Ct分别表示0 和t时的环氧化物浓度(mol/L),而C环状碳酸酯表示在指定时间生成的环状碳酸酯浓度(mol/L)。
2 结果与讨论
2.1 ILs/MOFs 复合材料表征
2.1.1 ILs/MOFs 复合材料结构分析
通过FT-IR 分别对TPA、DAIL 和DP 做了结构表征,结构如图5(a)所示。1695 cm-1处是TPA上C=O 键的伸缩振动峰,在TPA 和DAIL 反应之后,在1642 cm-1处出现新的C=N 伸缩振动峰[24],证明氨基和醛基成功组装。

图5 (a)TPA、DAIL、DP 的红外光谱谱图;(b)MIL-101、MIL-101-DAIL、MIL-101@DP-X 的红外光谱谱图Figure 5 (a) FT-IR spectra of TPA,DAIL and DP;(b) FT-IR spectra of MIL-101,MIL-101-DAIL and MIL-101@DP-X
MIL-101、MIL-101-DAIL 和 MIL-101@DP-X的FT-IR 光谱如图5(b)所示。1400 cm-1附近归属于MIL-101 骨架中C–C 拉伸振动特征峰,585 cm-1处是Cr–O 的特征峰,在747 和1015 cm-1附近的弱振动特征峰是MIL-101 骨架上苯环的C–H 面内弯曲振动特征峰[25]。结果表明,后合成修饰法构筑的MIL-101-DAIL、MIL-101@DP-X 保留了MIL-101 结构。除此之外,与MIL-101 相比,MIL-101@DP-X 和MIL-101-DAIL 中可以观察到1166 cm-1处归属于C–N 拉伸振动峰,1303 cm-1处归属于咪唑环拉伸振动峰[6],证明DP 和DAIL 成功负载于MIL-101 上。
2.1.2 晶体结构分析
粉末衍射(XRD)谱图(图6)显示了复合材料的晶体结构。由图6 可以看出,MIL-101 的XRD谱图与模拟谱图一致,且与之前报道(2θ=2.80°、3.30°、5.16°、5.60°、5.88°、8.46°、9.05°)[21,26]也 一致,表明MIL-101 成功合成且具有良好的结晶度。此外,对于样品MIL-101-DAIL、MIL-101@DP-1 和MIL-101@DP-2,主衍射峰的位置基本不变,表明MIL-101 在修饰后晶体结构保持完整[27]。

图6 MIL-101、MIL-101-DAIL、MIL-101@DP-X 的XRD 谱图Figure 6 Powder XRD patterns of MIL-101,MIL-101-DAIL and MIL-101@DP-X
2.1.3 比表面积和孔径分布
为了分析DP、DAIL 引入后复合材料的比表面积和孔结构变化,对其进行了N2吸附-脱附测试。如 图7(a)所 示,MIL-101、MIL-101-DAIL、MIL-101@DP-X 曲线为典型的I 型等温线[28]。
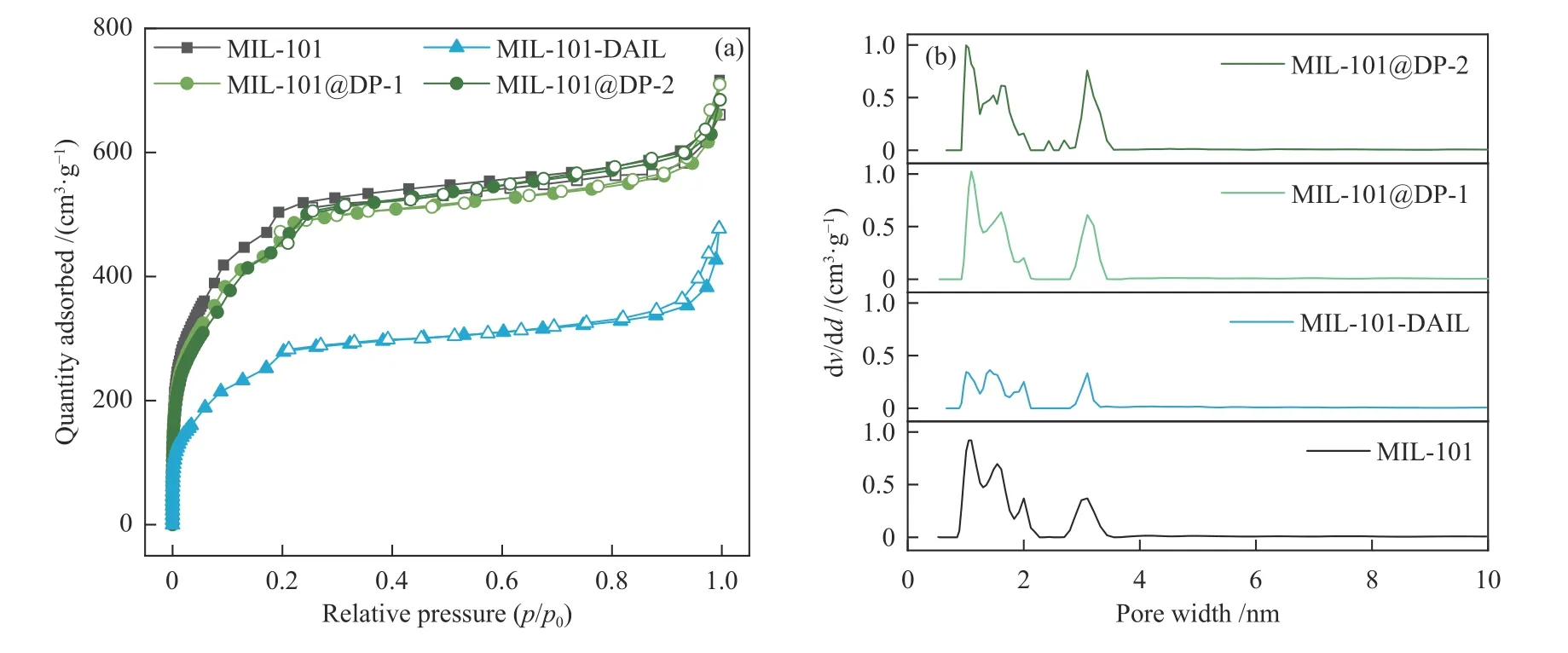
图7 N2 吸附-脱附等温曲线(a)和孔径分布(b)Figure 7 (a) N2 adsorption-desorption isotherms and (b) pore diameter distributions
表1 所示材料的比表面积和孔容分布表明,与MIL-101 相比,MIL-101-DAIL 的比表面积由1766 m2/g 下降到968 m2/g,孔容由1.07 cm3/g 下降到0.67 cm3/g,比表面积和孔容的显著降低表明DAIL 进入到MIL-101 孔道中。而MIL-101@DP-1 和MIL-101@DP-2 虽然比表面积分别降低至1618、1652 m2/g,但是下降程度很低。这是因为DAIL 与TPA 反应形成柔性的聚合物网络,聚合物网络与DAIL 单体相比体积变大,没有进入到MIL-101 孔道当中,因此,没有造成比表面积的大幅下降和孔容的降低。其中,比表面积少量下降可归因于有机材料在外表面的附着以及在干冷测量条件下聚合物在MIL-101 外表面坍塌并冻结,从而造成的部分孔堵塞[29]。在比表面积和孔容相近的情况下,MIL-101@DP-X 与MIL-101 相比,由于DAIL 的引入,具有了更多的催化位点,更有利于反应底物和CO2的吸附与扩散。

表1 MIL-101、MIL-101-DAIL、MIL-101@DP-X 的比表面积和孔容Table 1 Specific surface areas and pore volumes of MIL-101,MIL-101-DAIL and MIL-101@DP-X
2.1.4 形貌分析
为了观察聚合物DP 的引入对MIL-101 形貌的影响,对MIL-101、MIL-101@DP-2 做了SEM 表征,结果如图8 所示,DP 引入后,MIL-101 骨架的结构完整性保持不变。

图8 (a)、(b)MIL-101 的SEM 照片;(c)、(d)MIL-101@DP-2 的SEM 照片Figure 8 SEM images spectra of (a),(b) MIL-101,and (c),(d) MIL-101@DP-2
2.1.5 热稳定性分析
材料的热稳定性分析如图9 所示。对于MIL-101-DAIL 和MIL-101@DP-X,100 ℃之前的失重是材料在合成过程中使用的溶剂和孔道中吸附的水分。材料在100–250 ℃的失重主要是残留DMF 溶剂的释放和孔道内配位水分子的离开。在400 ℃时MIL-101@DP-X 的失重主要是由于表面固定的离子液体聚合物网络分解引起的[30]。在高于400 ℃的质量损失阶段归因于材料骨架的坍塌。所有材料在400 ℃之前保持稳定,在催化CO2与ECH 的环加成反应过程中可保持结构的稳定性。

图9 MIL-101、MIL-101-DAIL、MIL-101@DP-X 的DTG 曲线(a)和TGA 曲线(b)Figure 9 DTG (a) and TGA (b) curves of MIL-101,MIL-101-DAIL and MIL-101@DP-X
2.2 ILs/MOFs 复合材料催化性能评价
2.2.1 MIL-101、MIL-101-DAIL、MIL-101@DP-X催化性能比较
在0.1 MPa、50 ℃、24 h 的反应条件下,考察了MIL-101、MIL-101-DAIL 和MIL-101@DP-X 催化CO2/ECH 环加成反应性能。结果如图10 所示,在没有催化剂存在的情况下,没有观察到ECH 转化。当MIL-101 作为催化剂时,ECH 的转化率为50%,选择性为74%。根据以前文献报道[22,31],选择性低的是由于副产物二醇或者二聚体的生成,通过对催化后产物进行1H NMR 分析,确定在该反应当中副产物为3-氯丙烷-1,2-二醇。当DAIL 和DP 引入后转化率和选择性均有所提高,这是由于Cr3+、Cl-和咪唑基团的存在[13]。从图10 可以明显看出,MIL-101-DAIL 转化率和选择性均较MIL-101@DP 低,BET 结果表明,MIL-101-DAIL 的比表面积由1766 m2/g 下降到968 m2/g,孔容由1.07 cm3/g下降到0.67 cm3/g,虽然引入了亲核Cl-,但是比表面积和孔容的下降,不利于催化底物在孔道中的吸附扩散和反应。而MIL-101@DP-2结合了高比表面积(1652 m2/g)、Lewis酸位点Cr3+、亲核Cl-共同用于催化反应,在50 ℃下转化率为92%。

图10 MIL-101、MIL-101-DAIL、MIL-101@DP-X 在0.1 MPa、50 ℃、24 h 下的催化性能Figure 10 Catalytic performance of MIL-101,MIL-101-DAIL and MIL-101@DP-X at 0.1 MPa,50 ℃ for 24 h
2.2.2 反应温度对催化性能的影响
为了进一步研究MIL-101@DP-2 的催化性能,在0.1 MPa、24 h 的条件下,考察了温度对CO2/ECH环加成反应性能的影响,结果如图11 所示。ECH 的转化率随温度的升高而增加,当温度为80℃时,转化率为99%。这是由于CO2/ECH 环加成反应其固有活化能较高,需要热能输入来克服能量障碍[32]。另外从动力学角度考虑,在气液反应当中,传质系数会受到液体中气体扩散率(DL)和液体黏度(μL)的影响,温度升高会增加气体扩散、降低液体黏度,从而使气-液-固三相反应体系传质系数增大,加快反应进行[33],进而提高催化活性。

图11 MIL-101@DP-2 在0.1 MPa、24 h、不同温度下的催化性能Figure 11 Catalytic performance of MIL-101@DP-2 at 0.1 MPa,24 h and different temperatures
2.2.3 催化剂的循环稳定性能
非均相催化剂的循环稳定性对其实际应用具有重要意义,在反应条件为0.1 MPa、80 ℃、24 h下考察了MIL-101@DP-2 催化CO2/ECH 环加成反应的循环使用性能,结果如图12 所示,在经过四次循环后,MIL-101@DP-2 的催化活性出现下降。为了探究催化活性下降的原因,采用N2吸附-脱附、FT-IR 和XRD 对循环后的催化剂进行了测试(图13)。循环后MIL-101@DP-2 的FT-IR 谱图中仍存在1303、1120 cm-1咪唑上特征峰,表明在经过五次洗涤循环后DAIL 仍存在于复合材料中,具有一定的稳定性。然而同时在FT-IR 谱图中1798、1262 cm-1处出现了产物碳酸氯丙烯酯的C=O 和C–O 峰[34]。并且通过对循环后催化剂进行N2吸附-脱附测试发现了比表面积下降和孔隙堵塞,这造成了晶体结构破坏。从而导致催化性能下降[35]。

图12 MIL-101@DP-2 在0.1 MPa、80 ℃、24 h下的循环使用性能Figure 12 Cyclic performance of the MIL-101@DP-2 at 0.1 MPa,80 ℃ for 24 h
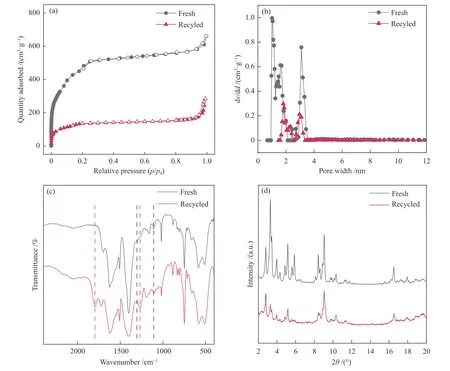
图13 MIL-101@DP-2 新鲜和回收后的(a)N2 吸附-脱附等温曲线,(b)孔径分布,(c)FT-IR 谱图,(d)XRD 谱图Figure 13 (a) N2 adsorption-desorption isotherms,(b) pore diameter distributions,(c) FT-IR spectra and(d) XRD patterns of fresh and recycled MIL-101@DP-2
2.2.4 催化剂的底物适用性
探究了MIL-101@DP-2 催化剂在常压、80 ℃、24 h 条件下对CO2与各种环氧化物环加成反应的普适性,结果如表2 所示。由于MIL-101@DP 具有的高比表面积和孔隙率,大尺寸的1,2-环氧己烷和1,2-环氧癸烷作为反应底物时转化率均达到99%。环氧环己烷作为反应底物时转化率为59%,这是因为环氧环己烷本身具有两个环,产生了空间位阻,更强的空间位阻减少了亲核攻击环氧环的机会,限制了开环[36]。对于氧化苯乙烯,由于苯环和环氧基之间的共轭效应导致了环氧化物β-C 中心的低反应性[37],转化率仅为29%。
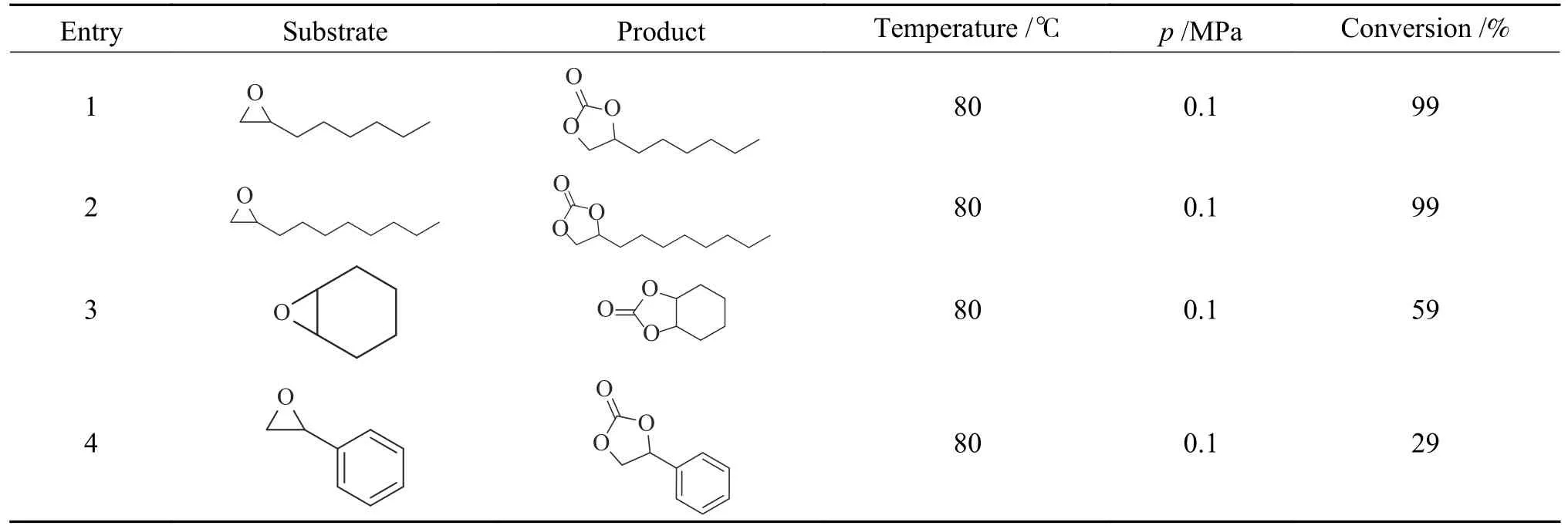
表2 MIL-101@DP-2 催化CO2 与各种环氧化合物合成碳酸盐Table 2 Synthetic carbonates from various epoxides catalyzed by MIL-101@DP-2
2.2.5 反应机理
根据实验结果和已有文献报道[13,38],提出了核壳结构的MIL-101@DP 在CO2与环氧化物环加成反应生成环状碳酸酯可能的机理(图14),首先环氧化物中的O 原子被MIL-101 上的Lewis 酸性Cr3+位点活化,激活环氧环,同时DP 壳层Cl–亲核攻击环氧化物上空间位阻较小的β-C,使环氧化物开环;同时咪唑环上叔氮吸附并活化CO2。随后活化的CO2被插入到开环的中间体中,最后Cl–离去,中间体合环生成环状碳酸酯,同时催化剂再生。
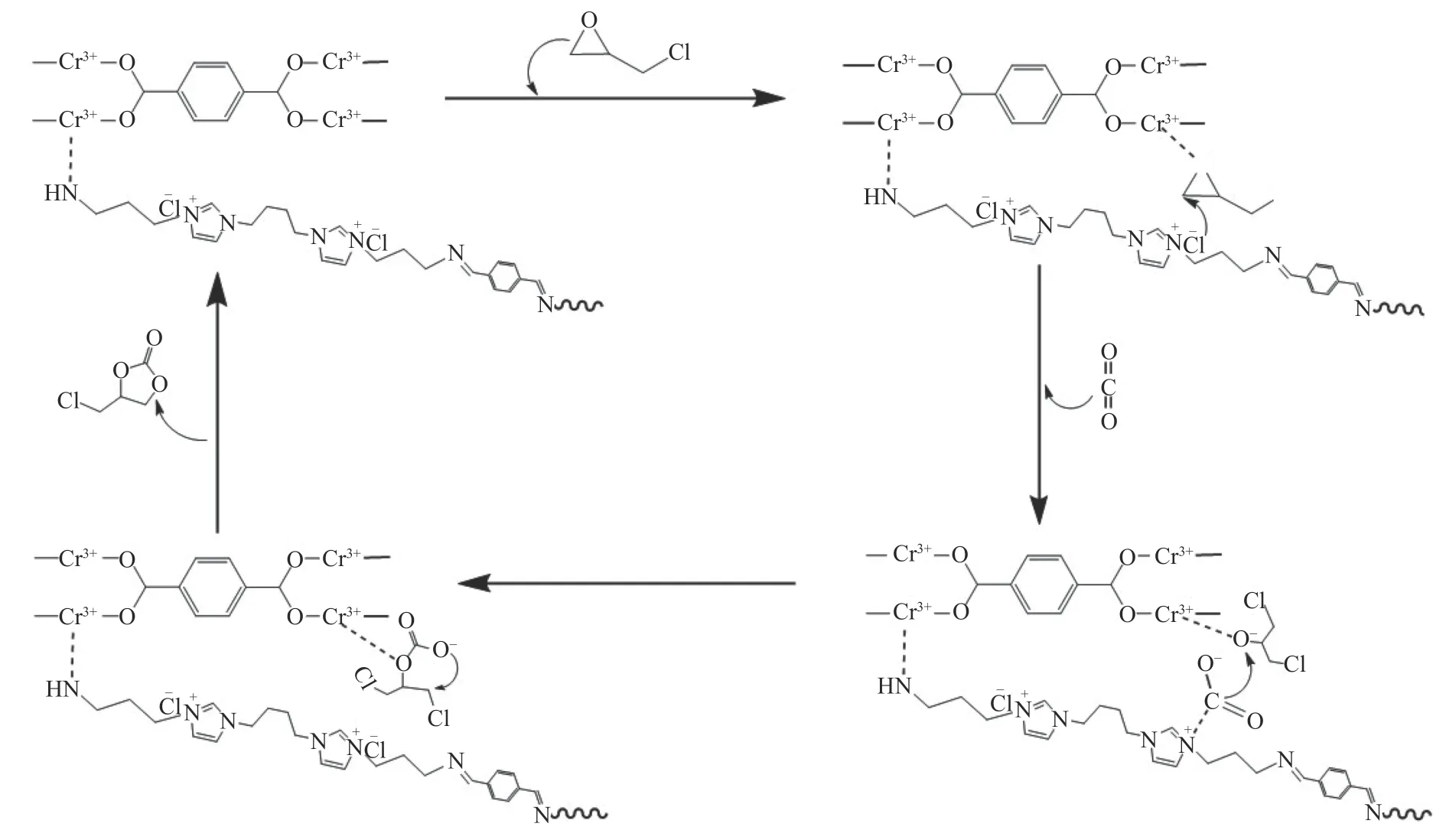
图14 MIL-101@DP 催化机理示意图Figure 14 Proposed catalytic reaction mechanism of MIL-101@DP
3 结论
采用后合成修饰的方法,制备了一种核壳结构的MOFs@ILs 复合材料(MIL-101@DP)用于高效催化CO2/ECH 环加成反应。MIL-101@DP保持了MIL-101(Cr)固有的高比表面积和高孔隙率,并通过离子液体成功引入亲核位点Cl–。由于Lewis 酸位点Cr3+与亲核位点Cl–的协同作用,MIL-101@DP 表现出了优异的催化活性,可在常压、80 ℃、24 h 且无助催化剂的条件下ECH 转化率可达99%。材料催化循环稳定性较好,有望成为有工业应用前景的环加成催化剂。本研究为定向设计酸碱协同高效催化剂提供了一种新的思路和方法。
