β-血红蛋白病的基因治疗临床试验进展*
2023-11-08刘倩宜陈晓琳刘思邈李欣瑜黄军就
刘倩宜, 陈晓琳, 刘思邈, 李欣瑜, 黄军就△
1中山大学生命科学学院基因功能与调控教育部重点实验室(广东广州 510275); 2中山大学孙逸仙纪念医院儿科血液专科(广东广州 510120)
β-血红蛋白病是一类由于编码β珠蛋白的β-珠蛋白基因(HBB)基因致病变异引起的疾病,包括β-地中海贫血症及镰状细胞病(sickle cell disease,SCD)等,是目前世界上最常见的单基因遗传病之一。在我国境内,β-血红蛋白病高发于南方地区,其中广东地区β-地中海贫血症基因携带率约为3.8%,粤北地区的β-地中海贫血症基因携带率可达6.34%[1-2]。β-血红蛋白病的治疗给患者、其家庭及社会均带来沉重的经济与医疗负担,因此亟需开发高效可行的β-血红蛋白病治愈性治疗方案。本文将针对β-血红蛋白病的基因治疗思路展开陈述,并对其存在问题与未来应用展开讨论。
1 血红蛋白及β-血红蛋白病
1.1 血红蛋白的组成 人类血红蛋白是由红细胞产生的含铁蛋白质,在机体内发挥运输氧气的功能。在胎儿时期至出生后早期,血红蛋白主要是由2条α-珠蛋白链及2条γ-珠蛋白链构成的胎儿血红蛋白四聚体(fetal hemoglobin,Hb F,α2γ2),而在出生后6~24个月内2条α-珠蛋白链及2条β-珠蛋白链构成的成人血红蛋白四聚体(adult hemoglobin,Hb A,α2β2)逐渐占据主导地位,此时人体内Hb F含量逐渐降低至约1%[3]。这一血红蛋白转换过程与11号染色体上β-珠蛋白基因簇的转录调控相关,其上线性排列的ε-珠蛋白基因(HBE1)、Gγ-珠蛋白基因(HBG2)、Aγ-珠蛋白基因(HBG1)、δ-珠蛋白基因(HBD)及HBB(图1)会随着胚胎发育及胎儿出生导致的个体生存环境中氧分压变化,在基因座控制区(locus control region,LCR)的调控下顺序转录,最终完成Hb F向Hb A的血红蛋白转换过程。
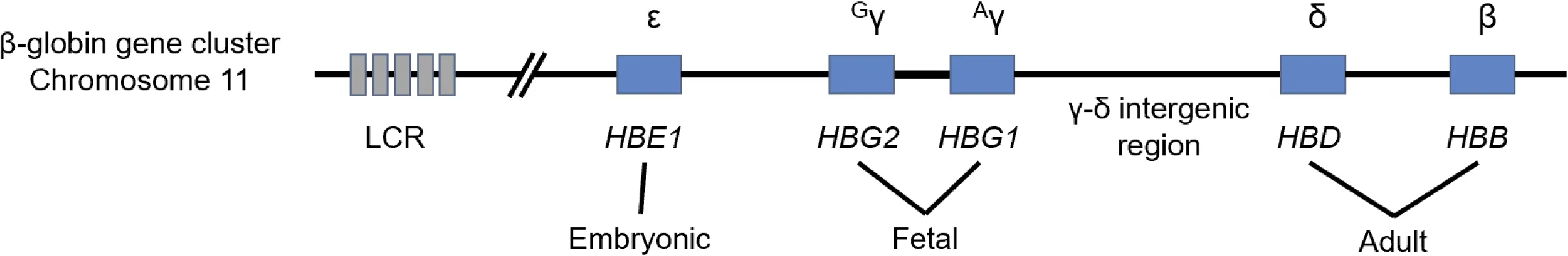
图1 β-珠蛋白基因簇上相关基因排列示意图
1.2 β-血红蛋白病的病理生理学 β-血红蛋白病主要是由β-珠蛋白基因HBB变异导致,根据突变后造成的病理生理现象可将其进一步分为SCD及β-地中海贫血症。其中SCD患者的HBB基因上第6位密码子从谷氨酸(GAG)突变为缬氨酸(GTG),导致镰状血红蛋白(sickle haemoglobin,HbS)生成。在脱氧状态下,HbS通过疏水键聚集形成多聚体,进而破坏红细胞结构,使其逐渐转变为镰状红细胞,并引起血管闭塞、溶血性贫血及血管病变。β-地中海贫血则是由HBB基因发生点突变、短片段插入或缺失所致,其引起患者β-珠蛋白链合成减少(β+)或缺失(β0),进而导致α-珠蛋白链与β-珠蛋白链合成失衡,冗余的α-珠蛋白链在红细胞及其前体细胞内发生自氧化并在细胞膜上沉积,造成细胞内氧化损伤及细胞膜结构破坏,最终导致机体发生溶血及无效造血。由于携带β-血红蛋白病相关突变的人群体内疟原虫生长受抑制,恶性疟疾进程缓解,所以HBB突变在疟疾暴露环境中经长期的自然选择而得到富集,最终呈现为β-血
红蛋白病与疟疾的流行地区在地理分布上的高度重叠。随着医疗水平的进步,除非洲地区外的大部分地区疟疾已得到控制,但β-血红蛋白病的地区性流行已成为全球医疗卫生的新负担。
1.3 β-血红蛋白病的常规治疗手段 目前,β-血红蛋白病的常规治疗策略主要集中在输血、药物治疗及造血干细胞(hematopoietic stem cells,HSCs)移植。输血治疗是临床上最为常用的治疗手段,但其需伴随除铁治疗,以避免输血及无效造血导致的铁过载损伤机体心脏、肝脏、内分泌器官等多种脏器。药物治疗通过诱导γ-珠蛋白基因表达抑制SCD患者中βS-珠蛋白聚集,缓解β-地中海贫血症患者中α-珠蛋白的冗余情况,从而缓解β-血红蛋白病临床表型。目前临床常用药物包括羟基脲、地西他滨、组蛋白脱乙酰酶抑制剂及沙利度胺等[4-6]。最近临床使用的罗特西普可通过促进红细胞晚期成熟降低β-地中海贫血患者的输血需求[7]。此外,通过减少镰状细胞脱水或镰化(GBT440),或针对SCD患者易发生血管闭塞现象(Crizanlizumab)开发的多种药物均已被证实具有临床治疗效果[8-9]。
与输血及药物治疗不同,HSCs移植是目前β-血红蛋白病仅有的治愈性治疗手段,但由于供体来源有限,通常只有少数具有人类白细胞抗原(human leukocyte antigen,HLA)相合同胞供体的患者有机会接受HSCs移植手术。对179例接受HLA相合同胞供体骨髓来源HSCs移植的重型β-地中海贫血症患者术后情况进行分析发现,移植后罹患急性或慢性移植物抗宿主病(graft-versus-host disease,GVHD)的患者比例达到了38%及13%[10]。移植过程中患者还需面临因免疫抑制引起的多种病毒、细菌及真菌的感染风险,以及因化疗导致的出血性膀胱炎等并发症引起的致死风险。HSCs移植手术通常耗费将近20万元人民币,其中近50%的费用用于购买与抑制免疫排斥及GVHD相关的药物,这也为患者家庭带来了沉重的经济负担。
2 基因治疗
基因治疗指将外源重组核酸转导至靶细胞中,利用核酸序列本身或其基因表达产物调节、修复、替换、添加或删除相关基因序列,从而达到疾病治疗等作用。其中针对单基因遗传病,基因治疗的策略主要包括基因替代、基因沉默及基因编辑。基因替代疗法主要通过外源提供具有正常功能的目的基因序列,弥补功能缺失的突变基因,适用于较小的隐形突变疾病治疗。基因沉默疗法则主要通过RNA干扰技术实现致病突变基因的沉默,适用于显性突变疾病的治疗。而基因编辑疗法是利用不同的基因编辑工具实现靶位点的基因序列插入、删除或替换,从而破坏、修复或激活目的基因的表达,以达到疾病治疗效果。
近年来,基因编辑工具的快速发展,为基因治疗的推进提供了有力的技术支持。现有基因编辑工具已从早期蛋白质介导靶序列识别的锌指核酸内切酶(zinc finger endonuclease,ZFN)及转录激活因子样效应核酸酶(transcription activator-like effector nuclease,TALEN),发展至RNA介导靶序列识别的CRISPR(clustered regularly interspaced palindromic repeats)/Cas(CRISPR-associated protein)系统。这些基因编辑工具介导DNA双链断裂(double-strand breaks,DSBs)后,断裂位点处发生非同源末端连接(nonhomologous end joining,NHEJ)介导的随机插入或缺失(insertions and deletions,indels)并引起相应靶序列的破坏,或在特定细胞周期中通过同源定向重组(homology directed repair,HDR)机制利用同源模板实现基因序列的精确修复或修改。
将失去或部分失去核酸酶活性的Cas蛋白(dCas或nCas)与胞嘧啶脱氨酶、腺苷脱氨酶或逆转录酶融合开发的胞嘧啶单碱基编辑器(cytidine base editor,CBE)、腺嘌呤单碱基编辑器(adenine base editor,ABE)及先导编辑器(prime editor,PE),可在不产生DSBs的情况下,实现精确的碱基编辑或短片段的插入、缺失或修改。这些新工具在丰富基因编辑类型的同时,减少了DSBs及NHEJ修复介导的副产物产生及相关细胞毒性[11-13]。Cas蛋白同系物或变体以其差异化的PAM识别序列及蛋白大小,进一步拓展了基因编辑疗法对不同致病突变的适用性,并优化了基因编辑工具在临床转化上的可操作性[14-17]。
2.1 β-血红蛋白病的基因治疗策略 β-血红蛋白病的基因治疗策略指对患者来源HSCs进行离体培养及基因修饰,以恢复其珠蛋白基因簇相关基因的正常表达,随后将修饰后的HSCs重新输注回患者体内。该策略不仅可以恢复患者体内血红蛋白的正常功能,还可以规避供体有限及GVHD等的限制(图2),为β-血红蛋白病的治愈性治疗手段的完善提供了可行的思路。β-血红蛋白病的基因治疗策略主要集中于基因替代疗法和基因编辑疗法,目前多项研究已进入临床试验阶段。
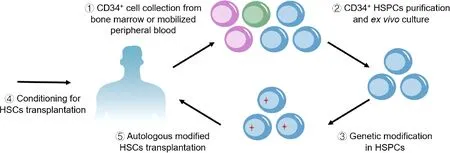
注:①从患者骨髓或动员外周血收集自体HSCs;②对CD34+ 造血干细胞及祖细胞(HSPCs)进行体外纯化及短期体外培养;③基于基因替代疗法或基因编辑疗法对纯化的CD34+ HSPCs进行体外基因修饰,使其恢复珠蛋白基因簇相关基因的正常表达;④在HSCs移植前,患者需接受由一种或多种化疗药物主导的预处理方案进行化疗;⑤在化疗结束后的1~2 d内,将修饰后的自体HSCs重新输注回患者体内,并在后期随访监测临床治疗效果
2.2 基因替代疗法 将具有正常功能的HBB基因转入β-血红蛋白病患者来源的HSCs以实现基因替代,能使红系细胞内β-珠蛋白表达水平升高,Hb A合成增多,从而达到治疗β-血红蛋白病的效果。相较于野生型β-珠蛋白,βA-T87Q-珠蛋白突变体具有更强的抑制HbS聚集及红细胞镰化的效果,可用于SCD的治疗。目前,蓝鸟公司已证实利用慢病毒载体递送βA-T87Q-珠蛋白编码序列至β-血红蛋白病患者来源的HSCs后进行自体造血干细胞移植的策略可行。SCD患者的临床试验结果显示,2/3的患者在接受治疗后的3.3~5.1年内维持稳定的βA-T87Q-珠蛋白表达水平,镰状红细胞比例降低,并摆脱了输血依赖性;1/3的患者降低了输血需求,但后续输血过程对βA-T87Q-珠蛋白表达存在抑制作用[18]。而β-地中海贫血症患者在治疗后15.7个月至6年时间内表现出输血依赖性降低,其中24/26例患者转变为非输血依赖性,且其无效红细胞生成及铁螯合症状均得到有效缓解[18-19]。2019年,基于βA-T87Q-珠蛋白基因替代疗法开发的Zynteglo(Bluebird)在欧盟有条件地获批上市,用于治疗输血依赖性β-地中海贫血症(transfusion-dependent β-thalassemia,TDT)。
国内多个研究团队也正在对该基因替代疗法的有效性及安全性进行验证。其中康霖生物于2022年5月公布的KL003临床试验结果显示,其较Zynteglo(Bluebird)缩短了移植后粒细胞及血小板植入时间至最长19 d,降低了治疗过程中的感染及出血风险。由中国人民解放军联勤保障部队第九二〇医院和上海本导基因合作开展的BD211临床试验(NCT05015920)也于2022年7月公布了1例β0/β0患者在治疗后3个月的随访期内保持血红蛋白水平的不断提升,初步摆脱了输血依赖性。目前,各项研究的后续随访工作仍在持续进行。
然而,目前针对该基因替代疗法的主要争议集中于与慢病毒整合活性相关的插入性肿瘤发生风险。Zynteglo(Bluebird)的HGB-206 A组(NCT02140554)临床试验中,2例SCD患者在接受治疗后分别被诊断出骨髓细胞异常增生症(myelodysplastic syndrome,MDS)及急性髓性白血病(acute myeloid leukemia,AML),致使Zynteglo的相关临床试验及在市销售被迫中断。后续Bluebird公司自行调查发现,MDS患者的恶性细胞中未发现慢病毒载体整合现象,而AML患者中慢病毒载体整合于VAMP4基因内含子上,但未发现该整合事件与AML发生存在相关性[20-21]。基于此,βA-T87Q-珠蛋白基因替代疗法已重新恢复临床研究。2022年8月17日,美国FDA宣布,批准Bluebird公司开发的基因疗法Zynteglo上市,用于治疗需要接受常规血红细胞输注的β-地中海贫血患者。Bluebird为Zynteglo的定价为280万美元,成为价格最昂贵的药物。但是,基于慢病毒载体递送功能性基因的疗法涉及的病毒整合活性相关的安全性问题、以及病毒载体在造血干细胞中的长期表达是否会被沉默等关键问题,仍有待长时间的随访研究。
2.3 基于基因编辑疗法诱导HBG表达 遗传性胎儿血红蛋白持续存在症(hereditary persistence of fetal hemoglobin,HPFH)指部分成人体内γ-珠蛋白未被沉默,因此Hb F仍维持高表达水平的现象。通常HPFH群体无明显临床症状,但对于同时患有β-血红蛋白病的患者而言,高水平的Hb F可抑制SCD患者体内的HbS聚集现象,并缓解β-地中海贫血症患者体内α-珠蛋白链与β-珠蛋白链间合成失衡情况。实际上,当Hb F表达水平达到20%以上即可有效缓解SCD患者的临床表型[22-23]。因此,利用基因编辑技术对HSCs进行精确的基因修饰以诱导HBG基因表达是治疗β-血红蛋白病的可行思路,其具体可通过降低HBG基因转录抑制因子BCL11A的表达,或模拟HPFH在HBG启动子区相关突变以减少BCL11A的结合抑制作用来实现(图3)。
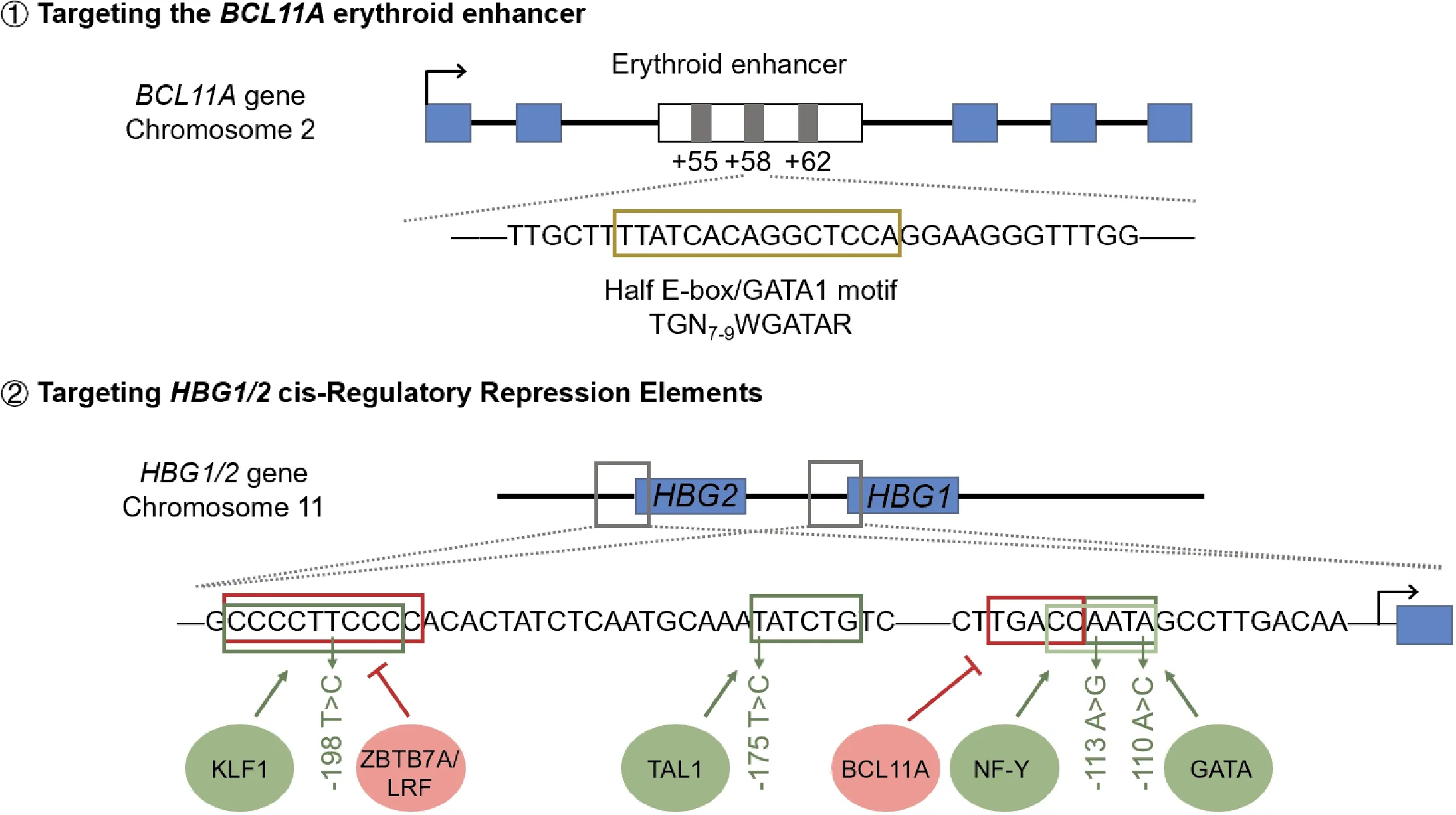
注:①棕色框则表示BCL11A+58红系增强子区核心基序,靶向破坏该基序可抑制BCL11A的表达,进而实现Hb F的激活;②图中红色框表示HBG1/2的转录抑制因子及其靶向结合基序,绿色框表示特定HPFH突变为相关转录激活因子提供新的结合位点,可激活Hb F的表达
2.3.1 抑制BCL11A表达以激活Hb F表达的治疗策略 BCL11A是HBG基因的关键转录抑制因子。敲低BCL11A可使原代成红细胞中γ-珠蛋白表达水平升高3.5~6.5倍,并使Hb F表达水平升高至23.6%~35.9%[24-25]。红系特异性敲低BCL11A的表达水平,可在不影响淋巴细胞分化成熟及HSCs移植后造血重建的同时,有效激活Hb F的表达。BCL11A序列中10 kb红系特异性增强子区域的发现为这一策略提供了可操作性。该区域中包含3个DNase Ⅰ超敏位点,根据其与BCL11A转录起始位点间的距离(kb)分别将其命名为+55、+58及+62。基于CRISPR/Cas9系统进行饱和诱变进一步确认灵长类动物中+58 区TGN7-9WGATAR Half E-box/GATA1结合基序是BCL11A增强子区的核心序列(图3),可作为红系特异性敲低BCL11A的理想靶点[26]。
现有研究已尝试利用ZFN及CRISPR/Cas9系统在BCL11A+58核心基序处诱导随机indels,以破坏其增强子活性,从而降低BCL11A表达水平,实现Hb F有效激活。其中基于ZFN系统开发的ST-400/BIVV003(Sangamo Therapeutics)已进入临床试验阶段。ST-400在TDT患者中的随访结果显示,在移植后5例受试患者体内Hb F表达水平最高可提升至(23.5±11.4)%,但在后续随访过程中(65~117周)回落至(7.7±4.1)%,患者未能摆脱输血依赖性[27]。而BIVV003在4例SCD患者中的临床试验则表现出了有效的临床治疗效果,移植后13周患者Hb F表达水平升高至15%~29%,所有患者均未出现血管闭塞危象[28]。基于CRISPR/Cas9系统靶向破坏BCL11A+58红系增强子的多项基因治疗临床研究也已得到初期结果。其中CTX001(CRISPR Therapeutics and Vertex Pharmaceuticals)的临床试验中,患者在移植后6个月HSPCs中可检测到的平均indels率均>70%[29-30]。其中TDT患者在移植后3个月体内总血红蛋白水平平均提升至>90 g/L,至最后一次随访时(移植后1.2~37.2个月)仅有2例TDT患者需再次接受输血治疗[29];而SCD患者在移植后3个月,Hb F表达水平提升至>20%,直至最后一次随访时(移植后2.0~32.3个月)未有患者出现血管闭塞性危象[30]。
我国多个研究团队也基于CRISPR/Cas9系统靶向破坏BCL11A+58增强子区的治疗策略展开了临床试验。其中华东师范大学吴宇轩团队、中南大学湘雅医院及上海邦耀联合发起的BRL-101临床试验(IIT)结果显示,2例TDT患者在移植自体HSPCs后45 d红细胞数量及总血红蛋白水平开始稳定增加,并在第75天左右达到健康水平[31];至移植后18个月时,患者Hb F水平达到142.7~158.8 g/L,并维持输血非依赖性及铁代谢情况的明显改善[31]。2022年11月,ET-01(博雅辑因)报告的临床试验结果中,1例β0/β+患者的接受ET-01输注后18个月,体内Hb F表达水平提升至89.87 g/L,总血红蛋白水平提升至110 g/L,在移植后87 d~15个月的时间内患者无需再次输血[32]。整体来看,基于CRISPR/Cas9系统在BCL11A+58位核心序列中诱导indels的策略在国内外的临床试验中均有效诱导Hb F表达水平提升,对β-血红蛋白病具有显著的治疗效果,并且表现出较高的安全性。
此外,利用单碱基编辑器在BCL11A+58位核心序列处诱导点突变也是抑制BCL11A表达的可行思路。在健康人群及β-地中海贫血症患者来源HSPCs中测试发现,ABE8e可在BCL11A+58位核心序列中的多个位点诱导高效的基因编辑,并使γ-珠蛋白表达水平升高至原来的4倍[33]。通过电穿孔递送A3A(N57Q)-BE3/sgRNA复合物至β-血红蛋白病患者来源的HSPCs中,也可实现86.3%~93.3%的碱基替代,并有效提高Hb F表达水平,具有一定临床转化价值[34]。
2.3.2 模拟HPFH在HBG启动子区相关突变以激活Hb F表达的治疗策略 HPFH群体中HBG基因启动子区的序列突变是其γ-珠蛋白持续表达的主要原因。其中,位于-115区及-200区附近的突变簇使得BCL11A和ZBTB7A等抑制蛋白不能有效结合,从而解除了这些蛋白对HBG的转录抑制作用;相反地,-110 A>C、-113 A>G、-175 T>C及-198 T>C突变为不同转录激活因子提供了新的结合位点,促进其对HBG的转录激活作用(图3)。相比之下,利用基因编辑技术在β-血红蛋白病患者HSPCs中诱导HBG启动子区发生天然存在的HPFH突变,有望在基因型及表型上更接近HPFH群体的临床实例,是激活Hb F表达更加安全且有效的策略。
基于这一思路开发的基因治疗药物EDIT-301(Editas Medicine)及RM-001(广州瑞风生物)已进入临床试验阶段[35-37]。两者分别通过CRISPR/Cas12a及CRISPR/Cas9系统在HBG启动子-115区CCAAT box处诱导indels以破坏BCL11A结合位点,激活Hb F表达。其中,EDIT-301在TDT患者动员外周血来源CD34+细胞中可诱导>80%的靶位点编辑,并改善TDT患者来源CD34+细胞红系体外成熟受阻情况[35]。广州瑞风生物开发的靶点药物RM-001在全球范围内率先在β0/β0患者中进行了临床试验。截至目前,该公司已经完成5例重症β地贫患者的IIT治疗,所有患者均在移植1个月后脱离输血[36-37]。已公布的随访数据中,患者在移植了经过修饰的自体HSCs后4个月Hb F表达量可达100 g/L,达到良好的临床治疗效果[36-37]。目前该疗法的IND(investigational new drug)申请也获得了中国国家药品监督管理局批准,正在开展临床试验。
利用单碱基编辑工具诱导HBG启动子区点突变是另外一种治疗策略。现有研究显示,利用hA3A-BE3/gRNA编辑系统可在-115C及-114C诱导约22% C>D(D包含A、T、G)的碱基取代效率,诱导<2%的indels,并未发现HBG2片段丢失现象[38]。对健康或患者来源的HSPCs进行相应编辑,证实该点突变可提高γ-珠蛋白表达水平至58.1%及44.2%[38]。新开发的eA3A-BE3及eeA3A-BE3系统可进一步提高碱基替代效率及后续γ-珠蛋白激活水平[39]。此外,近期研究利用ABE8e对β-地中海贫血症患者来源HSPCs中HBG-115区及BCL11A+58红系增强子区进行双重编辑,使体外诱导的红细胞中γ-珠蛋白表达水平进一步提高至约60%[33]。利用单碱基编辑工具模拟HBG-175 T>C点突变或-200区突变簇处点突变也被证实是可行的基因编辑治疗策略[40-41]。目前单碱基编辑策略还存在递送效率和安全性评估等临床转化所密切关注的问题。
2.4 基于基因编辑疗法精确修复HBB突变位点 尽管已有大量研究致力于通过药物或基因编辑手段诱导Hb F表达以治疗β-血红蛋白病,但相比于Hb A,Hb F更强的氧结合能力是否会引起治疗后患者组织缺氧仍未可知。因此,直接修复HBB基因突变是一种更理想的策略。
SCD的致病突变为Glu6Val,治疗靶点单一明确。基于CRISPR/Cas9系统及rAAV6递送的同源供体,可激活HSPCs中HDR修复通路以实现突变位点处的精确校正。但在未富集的情况下,该策略仅能实现约19%的基因修复,并产生约60%的随机indels,影响HBB基因的正常表达[42]。相比之下,利用ABE8e-NRCH将致病缬氨酸(GTG)转换为天然存在的非致病丙氨酸(GCG),可使SCD患者来源HSPCs红系分化后的βS-珠蛋白水平从(87±1.3)%降低至(17±3.0)%,细胞镰化率从47.7%降低至16.3%,具有一定的临床治疗意义[43]。
β-地中海贫血的致病突变类型繁多,针对不同的突变类型需建立不同的基因编辑策略。我国大陆地区β-地中海贫血症群体中6种主要的HBB基因突变类型为CD41/42(-TTCT)、CD17(A>T)、IVS-II-654(C>T)、-28(A>G)、CD26(G>A)、CD71/72(+A)。利用TALEN或CRISPR/Cas系统介导IVS-II-654(C>T)突变处产生随机indels,可破坏因突变造成的异常剪接位点,恢复HBBmRNA的正常剪接以达到临床治疗的效果[44-45]。基于CRISPR/Cas9与同源供体介导的HDR通路,研究者也在CD41/42(-TTCT)突变患者来源的人诱导多能性干细胞(hiPSCs)以及三原核胚胎中已实现了对该位点的校正[46-48]。但与SCD的研究结果类似,DSBs的修复过程常伴随明显的NHEJ修复通路偏好[46-48],并会影响基因治疗的精确性及有效性。
鉴于β-地中海贫血相关HBB突变多为点突变,利用单碱基编辑器或先导编辑器在靶位点处诱导精确的碱基修复同样具有可行性[49]。我国多个研究团队已针对大陆地区常见的β-地中海贫血症突变类型提出了有效的精确修复方案。其中,利用BE3 mRNA/sgRNA可在携带HBB-28(A>G)纯合突变的人核移植胚胎中介导7.0%~25.9%的碱基修复效率[50]。基于电穿孔递送的A3A(N57Q)-BE3/sgRNA复合体可进一步在患者动员外周血来源HSPCs中HBB-28位点介导68.2% C>T的修复效率[34]。利用ABE8e和ABE8e-SpRY也可使携带CD26(G>A)杂合突变的患者来源HSPCs中野生型基因频率从50.1%提升至90.7%及77.6%,并在小鼠实验中使正常β-血红蛋白水平提升至66.7%及76.3%[33]。此外,ABE8e-SpRY也可对携带IVS-Ⅱ-654(C>T)杂合突变患者来源HSPCs进行基因校正,使野生型基因频率从50.0%提升至77.9%,并使异常剪接mRNA比例从84.5%降低至36.3%[33]。不同PAM依赖性的单碱基编辑工具的开发及先导编辑器的出现,为更多HBB突变的精确修复提供了选择。
3 β-血红蛋白病的基因治疗策略的总结与展望
总的来说,基因治疗策略的发展为β-血红蛋白病的治疗提供了更多的选择。外源呈递βA-T87Q-珠蛋白编码序列或激活机体自身的γ-珠蛋白基因表达是β-血红蛋白病治疗的通用思路,目前相关研究较为成熟,并已有多项研究进入临床试验阶段;而对突变位点进行精确修复的策略则需根据具体突变类型进行不断地优化完善。考虑到研究的经济成本,尽管精确修复HBB突变的治疗理念充满吸引力,但对部分携带极罕见HBB突变的患者而言,采用β-血红蛋白病通用的基因治疗手段是更为可行的方案。而与基因替代疗法相比,基于基因编辑技术的治疗策略通过瞬转基因编辑工具实现靶位点编辑,可在不发生外源片段随机整合的情况下,通过改变HSCs的DNA序列实现长期的基因治疗效果,安全性相对更高。
在基因编辑工具的选择上,ZFN、TALEN或CRISPR/Cas系统均可在HSCs靶位点处诱导DSBs并进行NHEJ修复实现以较高的indels水平,在β-血红蛋白病的基因治疗策略中应用广泛,并已有多项相关研究进入临床试验阶段[51]。但与DSBs相关的p53通路的激活、基因组大片段丢失及染色体重排风险,以及随机indels带来的非预期基因编辑产物,均可能影响基因编辑的安全性及治疗效果[52-53]。新一代的基因编辑工具,如单碱基编辑器(ABE或CBE)及先导编辑器,可规避DSBs的产生并实现精确的基因编辑,从而降低靶位点处的indels水平,提高了基因编辑的精确性。但值得注意的是,受脱氨酶活性的影响,CBE系统在全基因组水平及全转录组水平上存在脱靶风险,其中部分研究发现脱靶位点在原癌基因及抑癌基因上[54-55]。同时,单碱基编辑器的编辑窗口内发生非预期编辑也会为碱基编辑修复带来副产物,影响基因治疗效果[56]。新开发的TadCBE系统基于TadA-8e进化得到了具有胞嘧啶脱氨酶活性的TadA-CD,其在保留靶位点处C>T高编辑效率的同时,有效降低了由脱氨酶引起的Cas9非依赖性脱靶效率[57]。基于此开发的TadCBE V106W系统进一步缩小了碱基编辑窗口,在编辑特异性上表现出了更好的应用前景[57]。但如何开发出更加系统科学的全基因组脱靶研究技术和分析方法仍是目前亟需解决的难点之一。此外,目前单碱基编辑工具在HSCs水平的递送效率有限,通过多次电穿孔递送RNP或以mRNA形式进行递送可在一定程度上提高递送效率,但多次电穿孔会造成明显的细胞损伤,mRNA的不稳定性也可能影响治疗结果的一致性[34,43]。因此,进一步优化基因编辑工具的递送策略也是目前β-血红蛋白病的基因编辑疗法的优化方向之一。
综上所述,现有临床试验结果展现了β-血红蛋白病基因治疗策略的显著疗效,为患者带来了重生的希望,但我们仍需通过长时间的随访来了解新疗法的安全性及药效时间等问题。
利益相关声明:论文内容不涉及相关利益冲突。
作者贡献说明:黄军就负责组织起草论文框架,提供写作思路,并对论文内容进行审阅和修订;刘倩宜负责文献查阅和整理,论文撰写;陈晓琳、刘思邈、李欣瑜负责论文的校阅修改。
