345例αβ复合型珠蛋白生成障碍性贫血血液学和基因型结果分析*
2015-03-16屈艳霞袁玉枝陈桂兰辜俊梅李志华左连东广州市人口和计划生育科学研究所5040广州市番禺区人口和计划生育技术服务站58000广州医科大学附属第三医院广州5050
屈艳霞,袁玉枝,杨 烨,江 帆,陈桂兰,辜俊梅,李志华,左连东(.广州市人口和计划生育科学研究所 5040;.广州市番禺区人口和计划生育技术服务站 58000;.广州医科大学附属第三医院,广州 5050)
·论 著·
345例αβ复合型珠蛋白生成障碍性贫血血液学和基因型结果分析*
屈艳霞1,袁玉枝1,杨 烨1,江 帆1,陈桂兰1,辜俊梅2,李志华3,左连东1
(1.广州市人口和计划生育科学研究所 510410;2.广州市番禺区人口和计划生育技术服务站 518000;3.广州医科大学附属第三医院,广州 510150)
目的 分析广州地区αβ复合型珠蛋白生成障碍性贫血的临床特征、基因突变类型,为临床遗传咨询提供依据。方法 进行血常规参数分析,筛查异常者进行高效液相色谱分析(HPLC)检测,应用跨越断裂点PCR(Gap-PCR)和PCR结合反向杂交(PCR-RDB)技术进行珠蛋白生成障碍性贫血基因分析。夫妇为同型珠蛋白生成障碍性贫血携带者进一步行胎儿珠蛋白生成障碍性贫血基因诊断,产后随访。结果αβ复合型珠蛋白生成障碍性贫血携带者的临床表现和HPLC结果,与单纯β-珠蛋白生成障碍性贫血杂合子携带者无差异;51个同型珠蛋白生成障碍性贫血家庭,经产前基因诊断检出重型β-珠蛋白生成障碍性贫血4例、重型α-珠蛋白生成障碍性贫血7例,均知情选择终止妊娠;产后随访结果均与产前诊断结果相符。结论 广州地区人群αβ复合型珠蛋白生成障碍性贫血携带率较高,β-珠蛋白生成障碍性贫血携带者同时进行α、β基因检测,对准确地进行遗传咨询和产前诊断具有重要意义。
α珠蛋白生成障碍性贫血;β珠蛋白生成障碍性贫血; 基因诊断; 产前诊断
珠蛋白生成障碍性贫血是一类由于珠蛋白基因缺失或缺陷使珠蛋白链合成受到部分或完全抑制而引起的一组遗传性溶血性贫血,高发于地中海地区、非洲、东南亚及我国西南、华南一带。据报道广东、广西、四川、贵州和台湾等地区人群中α-珠蛋白生成障碍性贫血发生率高达4.20%~17.55%,β-珠蛋白生成障碍性贫血为1.10%~6.43%[1]。在这些珠蛋白生成障碍性贫血高发区,αβ复合型珠蛋白生成障碍性贫血携带率亦较高,Li等[2]报道广东省人群中αβ复合型珠蛋白生成障碍性贫血的携带率为0.63%。为了解广州市αβ复合型珠蛋白生成障碍性贫血携带者的发生率、基因突变类型,并探讨预防重型珠蛋白生成障碍性贫血的有效手段及意义,总结广州市4个区2007年9月至2014年9月345例αβ复合型珠蛋白生成障碍性贫血携带者的基因诊断及产前基因诊断结果,进行分析如下。
1 资料与方法
1.1 研究对象 广州市黄埔区、番禺区、增城区、天河区通过广州市免费出生缺陷干预工程[3]共筛查64 200例,其中αβ复合型珠蛋白生成障碍性贫血携带者345例,男性170例,年龄23~39岁、平均(28.35±3.62)岁;女性175例,年龄21~37岁、平均(26.22±3.40)岁。345例αβ复合型珠蛋白生成障碍性贫血携带者根据基因检测结果又分为β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组和β复合血红蛋白H病(HbH病)组。同时选取200例(男女各100例)血液学表型正常、且经基因检测α-及β-珠蛋白生成障碍性贫血均未发现异常者为对照组,200例(男女各100例)经基因检测确诊仅携带β-珠蛋白生成障碍性贫血基因突变者作为β-珠蛋白生成障碍性贫血杂合子组。所有受试者在2007年9月至2013年2月完成筛查和产前诊断,2014年完成随访。所有受试者均知情同意。
1.2 方法
1.2.1 血常规初筛 取外周血2~4 mL,乙二胺四乙酸(EDTA)抗凝,应用五分类全自动血常规分析仪进行血液学检测,检测血红蛋白(Hb)、红细胞平均体积(MCV)、红细胞平均血红蛋白量(MCH)等。MCV≤82 fL和(或)MCH≤26 pg为筛查异常,将进行高效液相色谱分析(HPLC)检测。
1.2.2 HPLC分析 应用血红蛋白自动分析仪(Variant Ⅱ,BIO-RAD公司,USA)定量分析血红蛋白F(HbF)、血红蛋白A2(HbA2)等。 所有珠蛋白生成障碍性贫血初筛阳性的标本均检测α-珠蛋白生成障碍性贫血基因;HbA2≥3.5%和(或)HbF≥2.3%时,检测β-珠蛋白生成障碍性贫血基因。
1.2.3 基因型鉴定 采用Gap-PCR技术检测3种缺失型α-珠蛋白生成障碍性贫血基因突变类型(--SEA,-α3.7,-α4.2),采用PCR-RDB技术检测非缺失型α-珠蛋白生成障碍性贫血基因3种常见突变类型(αCS,αQS和αWS)和17种β-珠蛋白生成障碍性贫血常见突变类型(CD41-42、CD71-72、CD17、-28、CD26、IVS-Ⅱ-654、IVS-Ⅰ-1、IVS-Ⅰ-5、CD43、CD31、CD27/28、-32、-29、CD30、CD14-15、CAP和Int)。所有珠蛋白生成障碍性贫血基因诊断试剂盒购买于深圳益生堂生物技术有限公司。
1.2.4 产前基因诊断 对于夫妇为同型珠蛋白生成障碍性贫血基因携带者的孕妇,在其知情同意下统一转诊至广州医科大学附属第三医院。孕妇于术前进行遗传咨询,签署产前诊断相关手术知情同意书。在B超引导下,经孕妇腹壁行羊膜腔穿刺抽取羊水10 mL(孕18~24周)进行胎儿珠蛋白生成障碍性贫血基因诊断。
1.2.5 随访 根据胎儿珠蛋白生成障碍性贫血基因诊断结果,对所有受检者进行优生咨询和指导。随访所有受检者的妊娠结局,并于产后3~12个月对新生儿进行跟踪随访。
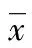
2 结 果
345例受检者经检测,其中β复合α珠蛋白生成障碍性贫血1组209例,β复合α珠蛋白生成障碍性贫血2组130例,β复合HbH病组6例。
2.1 血液学指标 345例受检者的血常规结果显示,此类患者多表现为小细胞、低Hb,MCV降低,MCH降低。Hb结果显示,此类患者临床表现较轻,贫血表现从轻度到正常不等,因此一般不会引起严重的临床后果。345例受检者的血常规初筛结果见表1。对照组、β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合HbH病组和β-珠蛋白生成障碍性贫血杂合子组之间Hb、MCV、MCH差异均有统计学意义(P<0.05);β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合血红蛋白H病(HbH病)组和β珠蛋白生成障碍性贫血杂合子组之间比较,MCV、MCH水平差异均有统计学意义(P<0.05),Hb水平差异无统计学意义(P>0.05);β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合HbH病组分别与β-珠蛋白生成障碍性贫血杂合子组进行比较,Hb水平差异无统计学意义(P>0.05),MCV、MCH水平差异均有统计学意义(P<0.05)。

表1 345例受检者的血液学指标结果分析±s)
2.2 HPLC结果 345例受检者中,除6例复合HbH患者血红蛋白电泳结果出现1.4%血红蛋白CS(HbCS),1.6%血红蛋白Bart′s(Hb Bart′s)外,其余339例均表现为HbA2升高,胎儿HbF正常或轻微升高(1.7%~4.2%)。345例受检者的HPLC结果分析见表2。对照组、β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合HbH病组和β-珠蛋白生成障碍性贫血杂合子组之间HbA2、HbF水平差异均有统计学意义(P<0.05);β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合HbH病组和β珠蛋白生成障碍性贫血杂合子组之间比较HbA2、HbF水平差异均无统计学意义(P>0.05),αβ复合型珠蛋白生成障碍性贫血与单纯β-珠蛋白生成障碍性贫血杂合子的血红蛋白电泳结果差异无统计学意义(P>0.05)。
表2 345例受检者的HPLC结果分析±s,%)
2.3 基因检测结果 345例受检者中,共检出11种β突变基因类型,分别为CD41-42、IVS-Ⅱ-654、-28、CD17、βE、CD71-72、-29、IVS-Ⅰ-1、CD27-28、CD43和CD14-15突变。其中CD41-42(-CTTT)移码突变频率最高,占47.54%(164/345)。共检出8种α突变,基因型分别为--SEA/αα、-α3.7/αα、-α4.2/αα、--SEA/-α3.7、--SEA/-α4.2、-α3.7/-α4.2、ααQS/αα和ααWS/αα,其中--SEA/αα最常见,占60.29%(208/345),6例为HbH病患者(包括4例--SEA/-α3.7和2例--SEA/-α4.2)外,其余均为α-珠蛋白生成障碍性贫血杂合子。345例受检者中,5对夫妇同为αβ复合型珠蛋白生成障碍性贫血携带者,32例配偶为α-珠蛋白生成障碍性贫血携带者,14例配偶为β-珠蛋白生成障碍性贫血携带者。345例受检者的基因检测结果见表3。
2.4 产前诊断及随访结果 5个同为αβ复合型珠蛋白生成障碍性贫血携带者的家庭,46对夫妇一方为αβ复合型珠蛋白生成障碍性贫血,其配偶为α-珠蛋白生成障碍性贫血或β-珠蛋白生成障碍性贫血携带者的家庭,均知情同意进行了胎儿珠蛋白生成障碍性贫血产前基因诊断,其中15例胎儿完全正常,25例为轻型珠蛋白生成障碍性贫血(其中轻型α-珠蛋白生成障碍性贫血14例、轻型β-珠蛋白生成障碍性贫血8例、轻型αβ复合型珠蛋白生成障碍性贫血3例),11例为重型珠蛋白生成障碍性贫血(重型β-珠蛋白生成障碍性贫血4例、重型α-珠蛋白生成障碍性贫血7例)。对所有受检者进行了遗传咨询,11例检出重型珠蛋白生成障碍性贫血胎儿的孕妇,在知情同意下选择了终止妊娠,其余40例均选择了继续妊娠。对所有受检者进行妊娠结局的随访,并于产后3、12个月对新生儿进行跟踪随访,结果均与产前基因诊断结果相符。
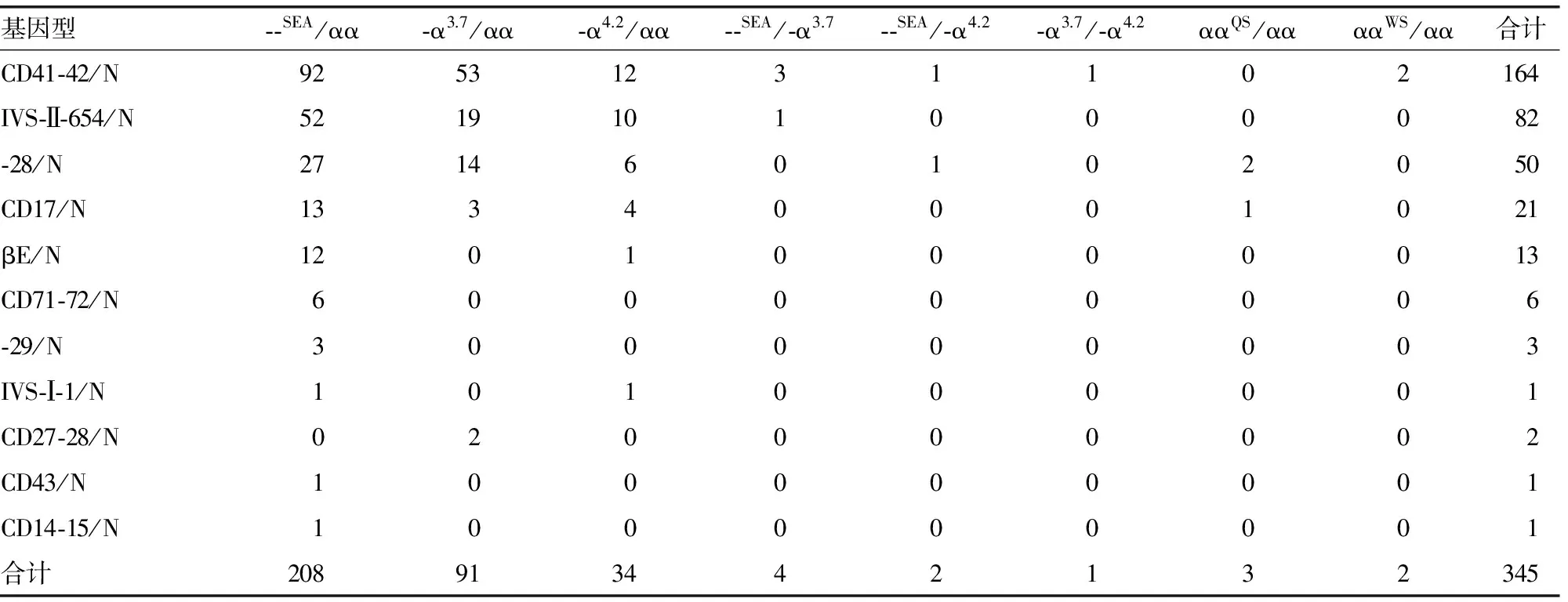
表3 345例αβ复合型珠蛋白生成障碍性贫血基因检测结果(n)
3 讨 论
珠蛋白生成障碍性贫血主要分为两种类型,分别是α-珠蛋白生成障碍性贫血和β-珠蛋白生成障碍性贫血,相同类型珠蛋白生成障碍性贫血杂合子间的婚配,生育重型珠蛋白生成障碍性贫血患儿的概率为1/4。不同类型珠蛋白生成障碍性贫血(α与β)杂合子婚配,没有生育重型珠蛋白生成障碍性贫血患儿的风险,但有生育αβ复合型珠蛋白生成障碍性贫血双重杂合子的可能。这种双重杂合子本身没有严重的临床表型,但无论与α或β杂合子婚配,均有可能生育重型珠蛋白生成障碍性贫血患儿。因此,提高αβ复合型珠蛋白生成障碍性贫血检出率,对指导珠蛋白生成障碍性贫血遗传咨询和准确进行产前诊断都具有重要意义。
从表型来看,所有αβ复合型珠蛋白生成障碍性贫血均表现出β-珠蛋白生成障碍性贫血杂合子的特点,即MCV和MCH低于健康人,HbA2增高,而α-珠蛋白生成障碍性贫血的特征被掩盖,与既往研究报道一致[4-5]。345例αβ复合型珠蛋白生成障碍性贫血与200例单纯β-珠蛋白生成障碍性贫血的血液学指标(MCV、MCH)和HPLC结果(HbA2、HbF)进行比较,差异均无统计学意义(P>0.05),说明血液学指标和电泳结果对αβ复合型珠蛋白生成障碍性贫血的准确检出作用都不大。故临床上αβ复合型珠蛋白生成障碍性贫血容易被漏检,且多误诊为β珠蛋白生成障碍性贫血,而αβ复合型珠蛋白生成障碍性贫血者与α-珠蛋白生成障碍性贫血或β-珠蛋白生成障碍性贫血杂合子个体婚配,均有可能生育出中、重型珠蛋白生成障碍性贫血儿。
αβ复合型珠蛋白生成障碍性贫血携带者比单纯β-珠蛋白生成障碍性贫血杂合子携带者的临床表现轻,本研究中β复合α珠蛋白生成障碍性贫血1组、β复合α珠蛋白生成障碍性贫血2组、β复合HbH病组分别与β-珠蛋白生成障碍性贫血杂合子组进行比较,MCV、MCH均值之间差异均有统计意义(P<0.05)。这是由于αβ复合型珠蛋白生成障碍性贫血者同时存在α及β珠蛋白基因缺陷,导致其α及β珠蛋白链的合成均相应减少,从而使β/α链比例失衡状态较单纯α珠蛋白生成障碍性贫血或单纯β珠蛋白生成障碍性贫血轻,以至不出现大量多余的β链,故其贫血程度也会有所减轻[6]。
从基因型来看,β-珠蛋白生成障碍性贫血发生的分子基础主要是由于β珠蛋白基因发生了突变,使β珠蛋白合成减少。β-珠蛋白生成障碍性贫血在我国南方以CD41-42、IVS-Ⅱ-654、-28、CD17、βE和CD71-72最为常见。在本研究中上述5种突变类型大约占β-珠蛋白生成障碍性贫血的97.38%,与以往的文献报道相符[7-8];α-珠蛋白生成障碍性贫血的分子基础则是α2和α1珠蛋白基因缺失或者发生点突变,在我国主要是东南亚缺失型α-珠蛋白生成障碍性贫血1(--SEA),α-珠蛋白生成障碍性贫血2则多为右侧缺失型(-α3.7)及左侧缺失型(-α4.2)。345例受检者中,以--SEA/αα最常见,占60.29%(208/345),与其他研究报道一致[9]。
345例受检者中,夫妇均为αβ复合型珠蛋白生成障碍性贫血携带者5对,配偶为α-珠蛋白生成障碍性贫血或β-珠蛋白生成障碍性贫血携带者46例。这51例孕妇均知情同意进行了胎儿珠蛋白生成障碍性贫血产前基因诊断,根据胎儿珠蛋白生成障碍性贫血基因诊断结果,对所有受检者进行了遗传咨询。11例检出为重型珠蛋白生成障碍性贫血胎儿的孕妇均知情选择了终止妊娠,有效地避免了这些重型珠蛋白生成障碍性贫血患儿的出生。对所有受检者进行妊娠结局的随访,并于产后3、12个月对新生儿进行跟踪随访,结果均与产前基因诊断结果相符。
广州市人群αβ复合型珠蛋白生成障碍性贫血的发生率较高,且缺乏特异性的血液学指标,只能通过基因分析确诊。因此,在临床工作中一定要加强对αβ复合型珠蛋白生成障碍性贫血的重视,尤其是对于配偶已确诊为α-珠蛋白生成障碍性贫血的情况下,应对β-珠蛋白生成障碍性贫血携带者同时进行α-珠蛋白生成障碍性贫血的基因检测,以便进行准确的遗传咨询和产前诊断,这对于避免中、重型珠蛋白生成障碍性贫血患儿的出生具有重要的意义。
[1]徐湘民.地中海贫血预防控制操作指南[M].北京:人民军医出版社,2011:28-29.
[2]Li B,Zhang XZ,Yin AH,et al.High prevance of thalassemia in migrant population in Guangdong province,China[J].BMC Public Health,2014,14:905-908.
[3]吴伟雄,冯善伟,江帆,等.广州市出生缺陷干预工程工作方案[S].广州:广州市人口和计划生育科学研究所,2013.
[4]李莉艳,李强,宋兰林,等.69例αβ复合型地中海贫血的血液学和基因型研究[J].实用妇产科杂志,2011,27(5):378-381.
[5]石青峰,杨峻,廖丽芬.αβ复合型地中海贫血的血液学和基因型特征[J].广西医学,2012,34(12):1670-1671.
[6]刘宁毅.复合型珠蛋白生成障碍性贫血的发生率及基因检测[J].检验医学与临床,2013,10(7):812-813.
[7]李东明,韦媛玉,晋武,等.13 610例孕妇地中海贫血筛查与产前诊断分析[J].中国妇幼保健,2014,29(15):2367-2369.
[8]韩俊英,曾瑞萍,胡彬,等.广东地区β地中海贫血复合缺失型地中海贫血双重杂合子检出率[J].中华血液学杂志,2001,22(10):514-516.
[9]王晶晶,朱文彪,黄霜,等.广州市1 381例育龄人群地中海贫血基因谱分析[J].中国优生与遗传,2015,23(2):5-7.
Analysis on hematology and genotypes results in 345 cases of αβ compound thalassemia*
QUYan-xia1,YUANYu-zhi1,YANGYe1,JIANGFan1,CHENGui-lan1,GUJun-mei2,LIZhi-hua3,ZUOLian-dong1
(1.GuangzhouMunicipalInstituteofPopulationandFamilyPlanning,Guangzhou,Guangdong510410,China;2.PanyuDistrictFamilyPlanningServiceStation,Guangzhou,Guangdong518000,China;3.ThirdAffiliatedHospitalofGuangzhouMedicalUniversity,Guangzhou,Guangdong510150,China)
Objective To investigate the clinical features and gene mutation types of αβ compound thalassemia to provide the basis for the clinical genetic counseling.Methods The blood routine parameters were analyzed.The individuals with abnormal results were detected by HPLC and the gene analysis of thalassemia was performed by using the gap-polymerase chain reaction(gap-PCR) and PCR combined with the reverse dot blot(PCR-RDB) technology.Husband and wife were the carriers of homotype thalassemia and further performed the gene diagnosis of fetal thalassemia and postpartum follow up.Results The clinical features and the HPLC results in the carriers of αβ compound thalassemia were similar to those of simple β-thalassemia heterozygous carriers.Among 51 families of homotype thalassemia,4 cases of severe β-thalassemia and 7 cases of severe α-thalassemia were detected by prenatal gene diagnosis.All cases selected the pregnancy termination after informing;the postpartum follow-up results were consistent with the prenatal diagnosis results.Conclusion The carrying rate of αβ compound thalassemia is higher among population in Guangzhou area.Simultaneously conducting the α and β gene detection in the β-thalassemia carriers has an important significance to accurately conduct the genetic counseling and prenatal diagnosis.
α-thalassemia; β-thalassemia; gene diagnosis; prenatal diagnosis
国家科技支撑计划资助项目(2006BAI05A02,2012BAI09B01);广东省计生委基金资助项目(2012208、20132031);广东省科技厅科技基础条件建设资助项目(2010B060100014)。
屈艳霞,女,助理研究员,硕士,研究方向是遗传与优生。
10.3969/j.issn.1672-9455.2015.22.003
A
1672-9455(2015)22-3297-03
2015-06-12
2015-09-14)
