端粒酶活性多重荧光定量PCR(Taqman探针)检测方法的建立及验证
2023-10-20董莉姜婷婷周宇荀李凯肖君华
董莉,姜婷婷,周宇荀,李凯,肖君华
东华大学化学化工与生物工程学院,上海201620
端粒酶催化亚基(telomerase reverse transcriptase,TERT)是端粒酶活性表达的关键组分,其编码的mRNA水平与端粒酶活性一致[1-2],因此,可以用TERT 的mRNA 水平来衡量端粒酶活性[3-4]。近年,细胞治疗及基因治疗得到快速发展[5],为更全面规范化控制细胞质量,可将端粒酶活性检测作为细胞成瘤性风险的评估标准,以提高细胞的生物安全性[6-7]。
目前,国内外有多种检测端粒酶活性的方法,如重复片段扩增(telomeric repeat amplification protocol,TRAP)法和TRAP-银染法[8-10],这些方法是通过聚丙烯酰胺凝胶电泳显示6 个碱基差异的条带来反映端粒酶活性,但由于步骤繁琐,成本高且存在放射性污染,已较少使用。另外,有厂家研发了SYBR Green实时荧光定量法,可间接测定端粒酶活性[11],但由于TERT基因表达量较低,导致Ct值波动性大,无法准确定量,且易受引物二聚体的干扰,使溶解曲线不稳定,结果存在一定的孔间差异和非特异性扩增,造成假阳性干扰[12-13]。因此,亟需建立一种具有高反应特异性、灵敏性及可对细胞基因进行精确定量的检测方法。本研究通过优化逆转录引物、TERT基因的定量引物和探针,并引入内参GAPDH基因,在单管中利用荧光定量PCR(Taqman 探针法)进行多重反应,提高反应特异性和灵敏性[14],同时验证方法的稳定性及重复性,以期应用于临床诊断和生物医药领域的评估检测。
1 材料与方法
1.1细胞株 293T和MRC-5细胞由中国科学院干细胞库提供;18 份正常间充质细胞样本和32 份乳腺癌细胞样本均由无锡翼和生物有限公司提供。
1.2主要试剂及仪器 Trizol 试剂购自日本TaKaRa公司;Probe qPCR Super Mix 购自苏州近岸蛋白科技有限公司;逆转录试剂盒Termo scientific Revert Aid First Strand cDNA Synthesis Kit及NanoDrop微量分光光度计购自美国Thermo公司;7500 Real Time PCR System购自美国Applied Biosystems公司。
1.3引物及探针设计 根据NCBI 中登录的TERT(7015)和GAPDH基因(2597)的CDS 序列,参照引物和探针的设计原则,应用Oligo 6.0 软件在CDS 序列3'末端设计特异性逆转录引物(RT-TERT 及RTGAPDH)。应用Snap gene 3.2.1软件设计1对GAPDH基因定量引物(GAPDH-1-F/R)和探针(GAPDH-1-P);设计TERT基因的2 对定量引物(TERT-1-F/R 及TERT-2-F/R)和FAM 荧光标记的探针(TERT-1-P及TERT-2-P),每对扩增产物长度约为200 bp。逆转录引物由美国Thermo 公司提供,定量引物及探针均由苏州泓迅生物科技有限公司合成。见表1。
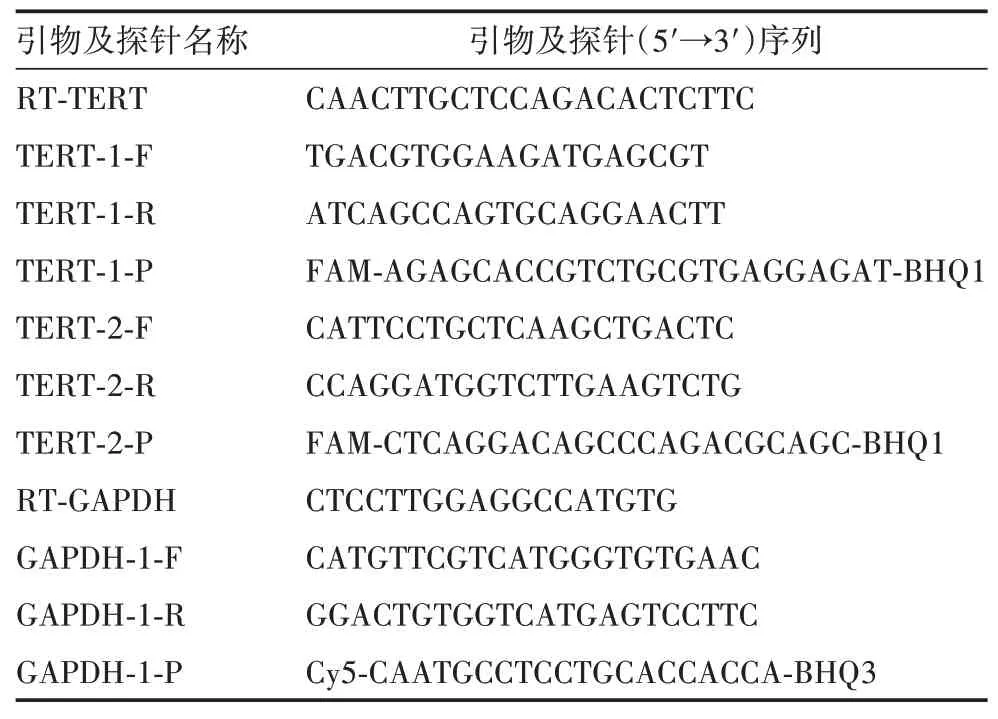
表1 TERT和GAPDH基因的引物及探针序列Tab.1 Primer and probe sequences of TERT and GAPDH genes
1.4方法的建立 荧光定量PCR 反应体系为:cDNA 2µL,Taq Man Multiplex qPCR buffer 10µL,ROX dye Ⅱ0.4µL,20µmol/L的TERT定量引物(F/R)各0.3µL,10µmol/L探针0.4µL,加入ddH2O至20µL。采用7500 Real Time PCR System 进行PCR扩增,扩增条件为:94 ℃5 min;95 ℃15 s,58 ℃1 min,72 ℃30 s,72 ℃采集荧光信号,共46个循环。
1.5荧光定量PCR 反应体系的优化 用Trizol 试剂提取MRC-5细胞RNA,在特异性逆转录引物(RT-TERT和RT-GAPDH)和随机引物作用下,通过逆转录获得两种荧光定量所需的cDNA;分别以两种cDNA 为模板进行多重荧光定量PCR 反应,方法同1.4 项。按定量引物的不同分为3 组:①TERT 第1 对定量引物及探针(TERT-1-F、TERT-1-R和TERT-1-P)与内参引物(GAPDH-1-F、GAPDH-1-R和GAPDH-1-P)作用进行双重荧光定量;②TERT第2对定量引物探针(TERT-2-F、TERT-2-R 和TERT-2-P)与内参引物(GAPDH-1-F、GAPDH-1-R 和GAPDH-1-P)作用进行双重荧光定量;③将TERT两对定量引物及探针混合后与内参引物进行3 重荧光定量(两种荧光)。以最小Ct值和较高荧光信号量(ΔRn)为最佳引物[15]。
1.6端粒酶阳性标准品及阴性标准品的制备 用Trizol
试剂提取293T 细胞RNA,逆转录为cDNA,将cDNA用超纯水进行2 倍稀释(初始浓度)后,再依次稀释为7.5、3.75、1.88、0.94和0.47 ng/µL 5个浓度,各取2 µL,采用优化方法进行检测,重复检测3 次,应用ABI 7500 软件生成标准曲线。另用Trizol 试剂提取MRC-5细胞RNA,相同方法制备端粒酶阴性标准品。
1.7方法的验证
1.7.1稳定性 取10 µL 端粒酶阳性标准品cDNA,加入等体积分数的TE(Tris-EDTA buffer solution)缓冲液,置50 ℃烘干过夜;加入20µL 的超纯水复溶,-20 ℃反复冻融3及5次,每次至少冻融12 h以上,取2 µL 作为模板,按优化方法进行检测,重复检测3次,以未冻融的样本为对照。比较反复冻融后的阳性标准品中TERT与GAPDH基因之间的△Ct,评价标准品的稳定性。
1.7.2精密性 以阳性标准品cDNA 为模板,采用优化方法进行检测,每个浓度平行检测3次,计算批内CV。将上述条件进行3次独立重复试验,计算批间CV。
1.8方法的应用 取19份正常间充质细胞样本和32份乳腺癌细胞样本,用Trizol 试剂提取细胞RNA,反转录为cDNA,以其为模板,采用优化方法进行检测,每个样本重复检测3次。
1.9统计学分析 应用GraphPad Prism 9软件进行统计学分析,试验数据以均值±标准差(±s)表示,组间比较采用t检验,以P<0.05为差异有统计学意义。
2 结果
2.1最佳荧光定量PCR 反应体系 ①组试验结果显示,在特异性逆转录引物和随机引物作用下,TERT基因均可获得较好的扩增曲线,但后者△Rn比前者低约1.5 倍,且TERT基因的Ct值约为25.2;②组试验结果与①组相似;③组试验结果显示,与随机引物比较,在特异性逆转录引物作用下,TERT基因的扩增效率和检测的灵敏度均大幅提高,且△Rn提高了约3 倍。见图1。因此,确定以特异性逆转录引物合成的cDNA 为模板,将TERT两对定量引物和探针混合后与GAPDH引物及探针进行多重荧光定量PCR(Taqman 探针法)检测,可大幅提高检测的灵敏度、特异性及TERT基因的荧光信号量。
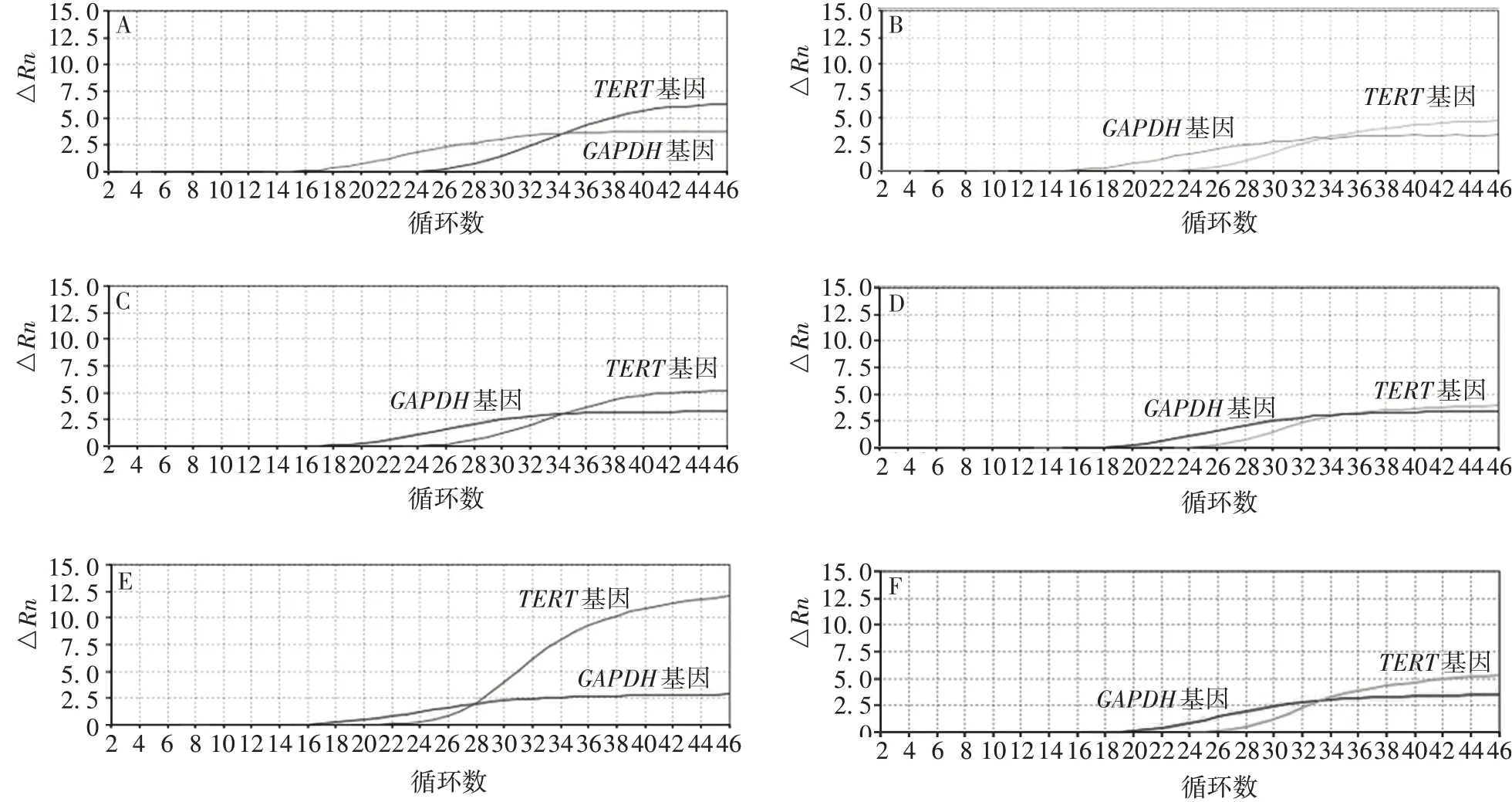
图1 荧光定量PCR反应体系的优化Fig.1 Optimization of fluorescence quantitative PCR reaction system
2.2阳性标准品的制备 5 个浓度梯度的阳性标准品TERT及GAPDH基因的Ct值分别为23~27 及15~19,TERT基因扩增曲线正常,且与GAPDH基因之间的△Ct保持稳定,见图2。确定制备标准品的端粒酶活性为阳性。且TERT和GAPDH基因的Ct值与样本量对数呈良好的线性关系,见图3。
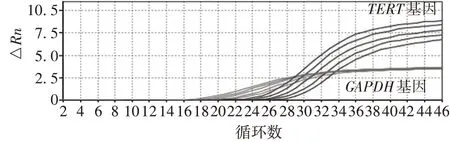
图2 阳性标准品TERT和GAPDH基因的扩增曲线Fig. 2 Amplification curves of positive standard TERT and GAPDH genes
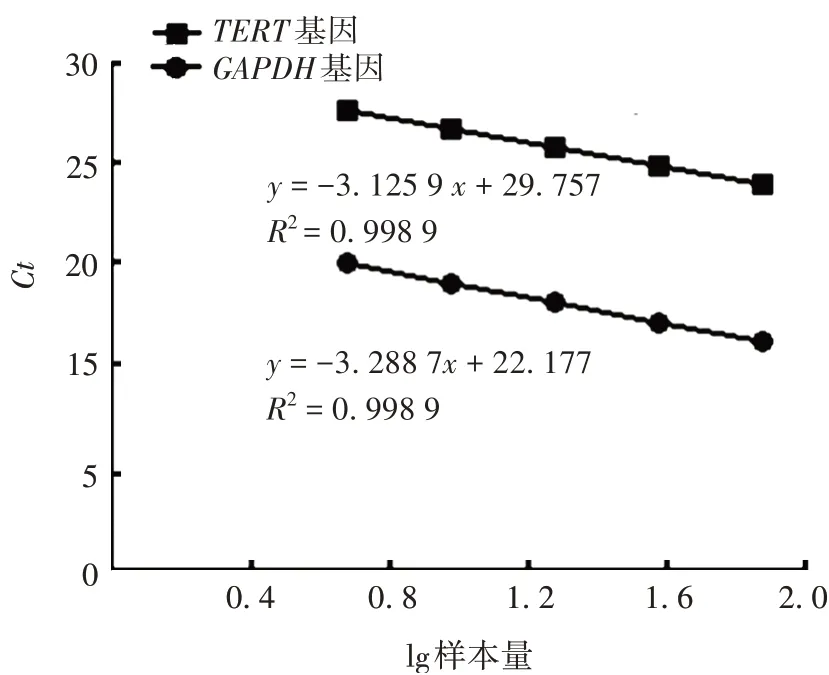
图3 阳性标准品TERT和GAPDH基因的标准曲线Fig.3 Standard curves of positive standard TERT and GAPDH genes
2.3阴性标准品的制备 5个浓度梯度的阴性标准品中,GAPDH基因扩增曲线正常,无TERT基因扩增曲线,且Ct值均>35,见图4。确定制备的阴性标准品的端粒酶活性为阴性。
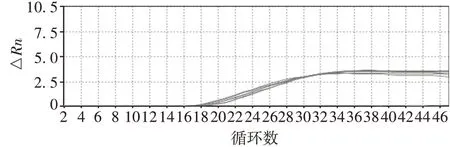
图4 阴性标准品GAPDH基因的扩增曲线Fig. 4 Amplification curves of negative standard GAPDH gene
2.4方法的验证
2.4.1稳定性 反复冻融3 及5 次阳性标准品中,TERT与GAPDH基因之间的△Ct与未冻融的阳性标准品比较,差异较小,△Ct均保持在8.0~8.6之间,见图5。表明该方法具有良好的稳定性。
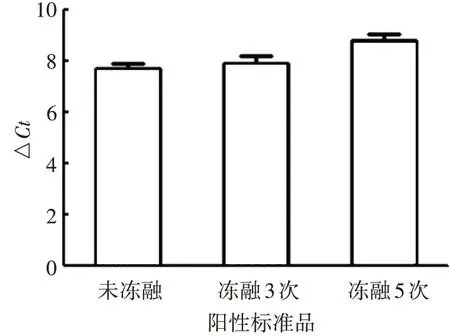
图5 稳定性验证结果Fig.5 Verification for stability
2.4.2精密性 以阳性标准品的cDNA为模板,Ct的批内CV为0.08%~0.75%,Ct的批间CV为0.12%~0.63%,均<1%,见表2。表明该方法具有良好的精密性。
表2 精密性验证结果(±s,n=3)Tab.2 Verification for precision(±s,n=3)
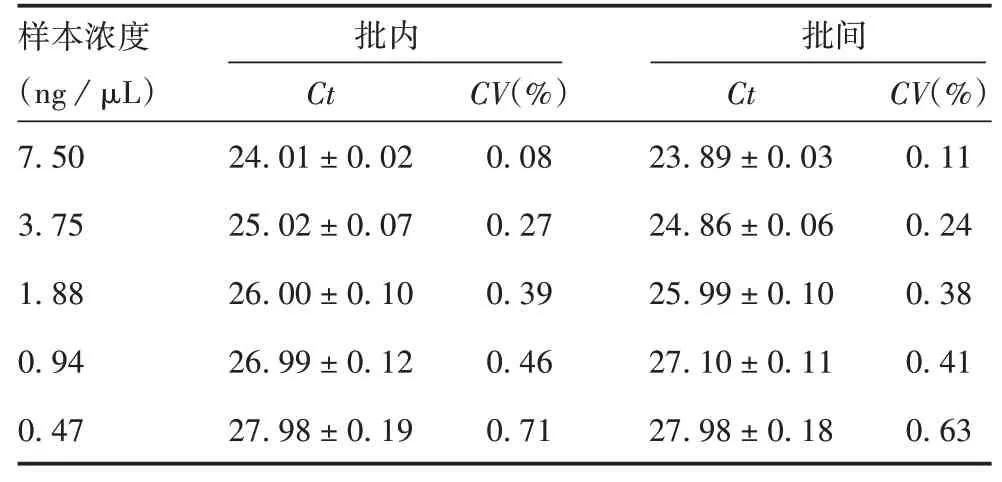
表2 精密性验证结果(±s,n=3)Tab.2 Verification for precision(±s,n=3)
?
2.5方法的应用 19 份正常间充质细胞样本TERT基因的Ct值均未检测出或>35,且GAPDH基因的Ct平均值为18,表明端粒酶活性均为阴性;32 份乳腺癌细胞样本TERT基因Ct值均<35,有良好的扩增曲线,表明端粒酶活性均为阳性。见图6。且TERT与GAPDH基因之间的△Ct保持稳定,符合预期要求。19 份正常间充质细胞样本与32 份癌细胞样本的TERT基因的Ct值差异有统计学意义(t=4.236,P<0.001),GAPDH基因的Ct值差异无统计学意义(t=0.597,P>0.05)。见图7。
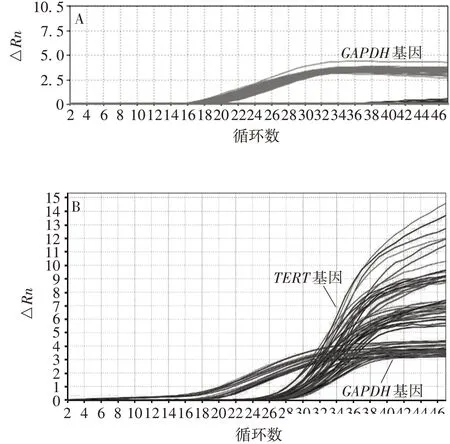
图6 间充质细胞(A)及乳腺癌细胞样本(B)的扩增曲线Fig. 6 Amplification curves of mesenchymal cell(A)and breast cancer cell samples(B)
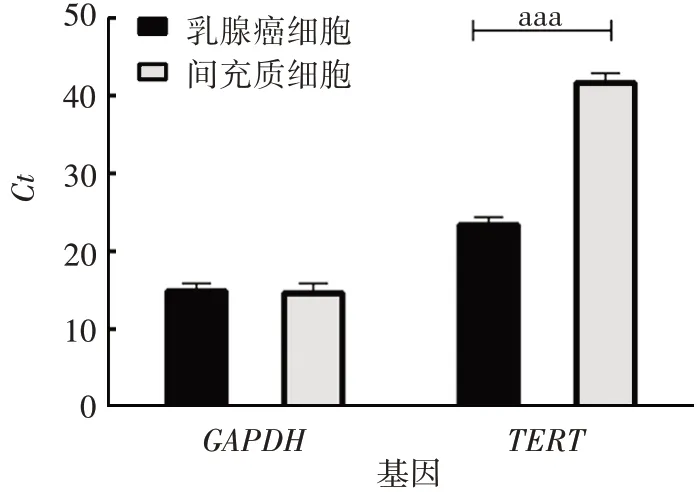
图7 间充质细胞及乳腺癌细胞样本TERT 及GAPDH 基因的Ct值Fig. 7 Ct values of TERT and GAPDH genes in mesenchymal cell and breast cancer cell samples
3 讨 论
常用于分析端粒长度的细胞类型主要是血淋巴细胞和其他单核细胞,通常称为外周血单核细胞(peripheral blood mononuclear cell,PBMC)[16],这些细胞被激活时能够上调端粒酶活性。肿瘤发生过程中,端粒酶活性高度上调,在大多肿瘤组织中,端粒酶活性较高。这种活性升高的状态使端粒长度得以维持,即使在肿瘤细胞中端粒长度较短,也能使细胞无限期生长[17-18]。因此,通过检测端粒酶活性可快速判断正常细胞是否转变为肿瘤细胞,可用于早期肿瘤的诊断。
MRC-5 细胞是14 周龄男性胎儿正常肺细胞,免疫原性好,有良好的耐受性,可选作阴性标准品[19]。由于TERT在正常细胞中的表达量较低,使端粒酶活性检测变得困难,为解决该问题,有研究建立了一种荧光定量PCR 与TRAP 结合的检测方法,用该方法检测13例淋巴瘤样本,虽然比TRAP法灵敏度高9倍,但无法避免延伸引物的二聚体干扰和TRAP法带来的放射性污染,且该方法的精密性验证CV约为5%[20-21]。本研究对TERT基因的定量引物和探针进行了优化,将2对同一种荧光的探针进行合并后与GAPDH基因在单管中利用Taqman 探针法进行多重定量PCR 反应,不仅安全高效,还在一定程度上避免了孔间差异和产物非特异性扩增,且CV均<1%,更能稳定、灵敏地检测端粒酶活性。OU等[22]为避免TRAP法扩增后的繁琐步骤(如聚丙烯酰胺凝胶电泳),设计了一种基于氧化石墨烯的核酸探针检测法,操作相对简单,但无法确保样本在不同批次检测结果的一致性和稳定性,不利于大量临床样本的检测。本研究将标准品反复冻融,采用建立方法进行检测,结果显示,TERT基因的扩增曲线正常,且与GAPDH基因的△Ct保持恒定,证明该方法具有良好的稳定性。YANG 等[23]利用TRAP 法对167 份胃黏膜样本进行端粒酶活性检测,结果显示,65份胃癌样本中阳性率达92.3%,57 份慢性萎缩性胃炎胃黏膜样本中阳性率为24.6%,45份正常胃黏膜样本中阴性率为100%。本研究检测的51 份样本中,乳腺癌细胞与正常间充质细胞中TERT与GAPDH基因之间的相对表达水平差异有统计学意义(P<0.001),与相关文献报道癌细胞中具有较高端粒酶活性相符[18,24-25]。
综上所述,本研究建立的多重荧光定量PCR(Taqman 探针)法可稳定高效检测端粒酶活性,一定程度上改变了TERT基因的荧光信号量,提高了检测灵敏度。由于85%~98%的肿瘤细胞均存在端粒酶活性[25],通过对端粒酶活性的筛查,可快速预判癌变风险。另外,端粒酶还可作为抗肿瘤的重要靶点[26],有望成为未来生物医学和临床医学新的研究方向。
