芳基四硫富瓦烯与碘电荷转移复合物的合成及其晶体结构
2023-10-19马龙飞
左 琦 马龙飞
(河南警察学院刑事科学技术系,郑州 450046)
电子给体和电子受体之间的电荷转移在超分子组装和超导材料的合成中起着重要作用[1-4]。有机分子结构和电子态的多样性,对实现复合物电子态和堆积结构的多样性具有重要作用,进而会对复合物光学性质、电学性质、磁学性质等产生重要影响[5]。
碘能够形成稳定的高聚碘阴离子[6-10]。高聚碘阴离子以其良好的物理化学性能(如高导电性和氧化还原能力)已经引起科学家们的广泛关注[11-16]。高聚碘阴离子主要由3 种基本的堆积单元组成:I2、I-和I3-,它们之间通过I 与I 的范德瓦耳斯力相互连接,可以呈现出多种结构:1D 链状、2D 和3D 网格状结构[6-10]。作为补偿离子的阳离子对高聚碘阴离子形成不同的结构具有重要影响[8]。四硫富瓦烯(tetrathiafulvalene,TTF)及其衍生物作为良好的电子给体具有3 种电化学可逆状态:(TTF)0、(TTF)+·和(TTF)2+[17]。由于其独特的电子和结构特性,TTF 及其衍生物作为重要的有机功能材料引起了人们的广泛关注[18-19]。硫原子桥联芳基取代四硫富瓦烯(Ar-S-TTF)作为良好的电子给体,其外围芳基可以围绕C—S 键自由旋转,可以与客体分子间相互作用,形成独特的分子堆积结构。Ar-S-TTF 多变的分子构型及其极化性对聚碘阴离子长度和几何形状会产生重要影响,进而会对电荷转移复合物的物理化学性能产生影响。
本文中,我们报道了Ar-S-TTF 化合物1~3(图1)与碘的电荷转移复合物的合成、电荷转移和晶体结构。研究了Ar-S-TTF 和聚碘阴离子间分子构型和电荷转移的相互影响。
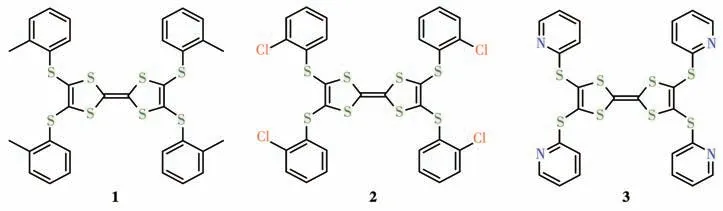
图1 化合物1~3的分子结构Fig.1 Molecular structure of compounds 1-3
1 实验部分
1.1 试剂与仪器
所用溶剂均按标准方法进行处理[20]。碘购自天津市科密欧试剂公司,纯度98%。
电化学性质在RST5000 电化学工作站上进行测试。测试条件为溶剂:二氯甲烷;温度:20 ℃;浓度:5×10-4mol·L-1;支持电解质:TBAPF6(四丁基六氟磷酸铵);扫描速率:0.05 V·s-1;工作电极:铂碳电极;辅助电极:Pt 电极;参比电极:饱和甘汞电极。UVVis 吸收光谱在Shimadzu UV-2600 紫外可见分光光度计上进行测试。
1.2 电荷转移复合物的合成
化合物1~3 根据已报道的方法进行合成[21],使用前进行重结晶,其氧化还原电位E1/2,A和E1/2,B如表1 所示。氧化还原电位对电荷转移复合物的合成以及复合物中电荷转移具有重要影响。通过对比我们可以发现芳基上邻位取代基由甲基变为氯原子会对化合物电化学性质产生影响。而化合物1和化合物3的氧化还原电位变化不明显。
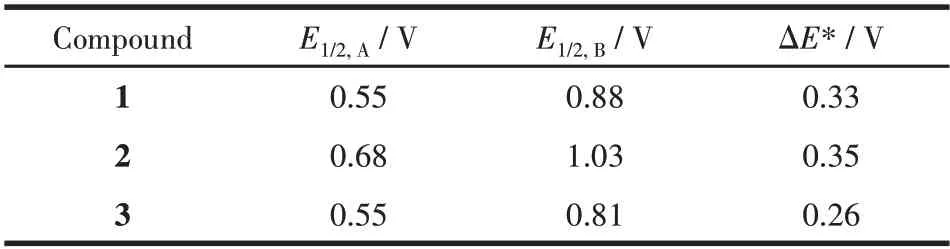
表1 化合物1~3的电化学数据Table 1 Electrochemical data of compounds 1-3
化合物1~3与碘合成复合物的方法相似,以(1+·)(I3)·I2的合成为例说明如下。将含有6.9 mg(10 μmol)化合物1 的二氯甲烷溶液(重蒸)5 mL 置于20mL 玻璃试管中,缓慢加入2 mL 二氯甲烷和2 mL 正己烷作为隔层,然后加入含15.2 mg(60 μmol)碘的正己烷溶液(重蒸)5 mL,用封口膜封口后避光放置。1周后,将封口膜用针刺破,缓慢挥发溶剂。1周后,用正己烷洗涤、过滤、干燥,得6 mg 黑色块状晶体。所有复合物的合成方法、组成、产量和外观列于表2。

表2 复合物的合成条件、组成、产量和外观Table 2 Preparation condition,composition,yield,and appearance of the complexes
1.3 晶体结构的测定
选取大小合适、外观良好的单晶样品,用SuperNova X 射线衍射仪(Agilent)进行测试,晶体结构采用直接法由Olex2[22]解析,并通过SHELXL-97[23]进行校正。非氢原子的坐标及各向异性热参数用全矩阵最小二乘法进行最后修正,氢原子坐标由理论加氢获得。晶体数据列于表3中。
表3 复合物的晶体学数据Table 3 Crystallographic data for the complexes
CCDC:2258351,(1+·)(I3)·I2;2258352,(2+·)(I5)·I2;2258353,(32+)(I3)2。
2 结果与讨论
2.1 紫外可见吸收光谱
室温条件下,测试了10 μmol·L-1(CH2Cl2为溶剂)的化合物1~3 加入碘后的紫外可见吸收光谱,结果如图2所示。可以观察到随着碘的滴加,化合物1和3 在700~1 000 nm 处产生了新的吸收峰,这是TTF自由基离子的特征吸收峰[24-25],说明在溶液中化合物1 和3 与碘发生了电荷转移,化合物1 和3 被氧化为阳离子自由基。而化合物2随着碘的加入并未产生新的特征吸收峰,说明化合物2 在溶液中并没有被氧化,这与化合物2的氧化还原电位有关。
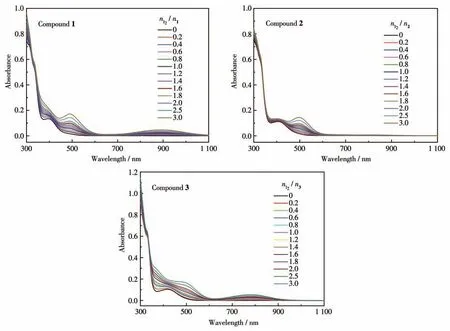
图2 化合物1~3的溶液在逐渐加入I2时的UV-Vis吸收光谱图Fig.2 UV-Vis absorption spectra of the solutions of compounds 1-3 upon the gradual addition of I2
2.2 复合物的电荷转移
TTF 衍生物自身所带电荷的变化对中心TTF 骨架键长具有明显影响。因此,复合物中TTF 衍生的价态可以按照Day 等的经验公式[26],根据键长进行估算。ρ=6.347-74.36δ,而δ=(b+c)-(a+d),其中δ为每个电荷状态之间给出最大区别和最小偏差的参数;a、b、c和d分别代表中心TTF骨架C=C键、C—S键、C—S 键和C—C 键的平均键长。复合物(1+·)(I3)·I2、(2+·)(I5)·I2和(32+)(I3)2中TTF衍生物骨架键长和所带电荷如表4 所示。化合物1 在复合物(1+·)(I3)·I2中显示+1 价,这与溶液中碘滴定化合物1 时电荷转移情况一致。化合物2 在复合物中也显示+1 价,这与其在溶液中无电荷转移是不一致的。化合物3在复合物中显示+2 价,这与溶液中化合物3 被氧化为阳离子自由基也不一致。化合物1~3与碘之间的电荷转移与化合物的氧化还原电位以及复合物的堆积结构有密切关系。下面我们将对分子的几何结构和复合物堆积结构进行讨论。
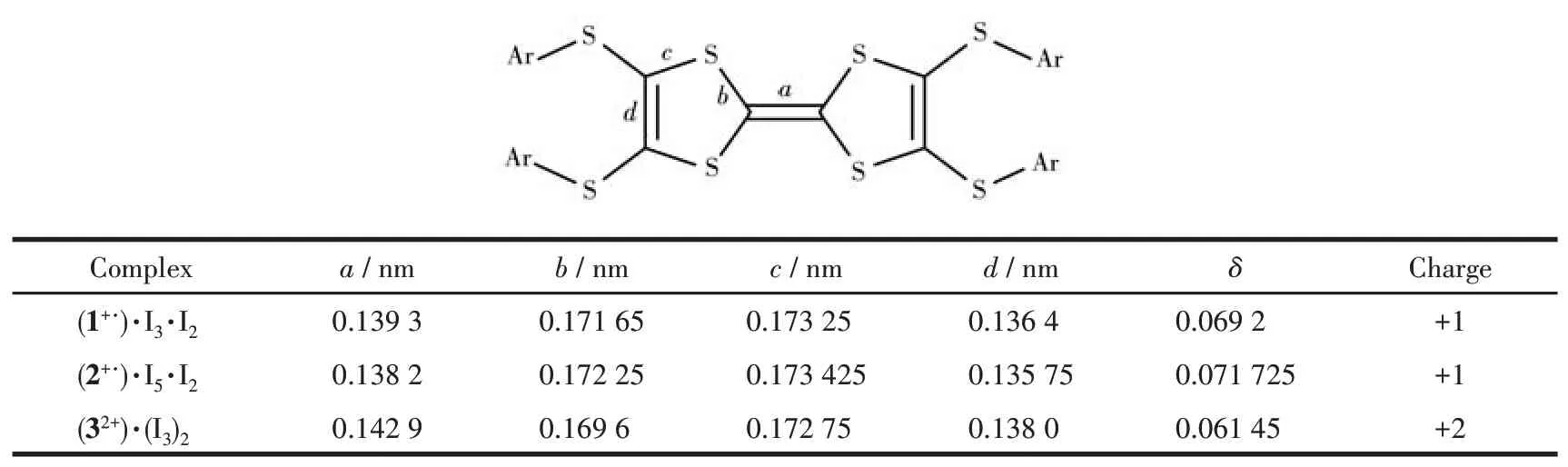
表4 复合物中心TTF骨架的键长和所带电荷Table 4 Selected bond lengths of the central TTF cores and calculated charge in the complexes
2.3 复合物(1+·)(I3)·I2的晶体结构
复合物(1+·)(I3)·I2的晶体属于单斜晶系C2/c 空间群。不对称单元中,包括半个1+·、半个I3-阴离子和半个I2。如图3 所示,复合物中1+·呈椅式构型,中心C=C 键长为0.139 nm,理论计算δ为0.069 2,依据Day等的经验公式,化合物1在复合物中显+1价。沿a轴方向,1+·呈柱状堆积,柱内分子间通过芳基碳原子与外围硫原子间的2 个S…C(0.340 nm)发生相互作用。柱间通过甲基氢原子之间的H…H(0.339 nm)产生相互作用,1+·呈二维网格状堆积。如图3b所示,I2i—I1—I2ii通过共价键连接形成I3-负离子,I1—I2ii键长和I2i—I1 键长均为0.292 nm[27-29]。I3i—I3ii(0.275 nm)与I2(0.272 nm)键长一致,所以复合物晶体中I3i—I3ii以I2构型存在[27-29]。相邻的I3-离子和I2之间通过I…I(0.345 nm)范德瓦耳斯力相互连接。和I2交替连接沿c轴方向呈无限延伸的一维链状结构,如图3b所示。每个1+·通过2个I…S(0.375 nm)与2个I2相互作用,1+·与I3-之间没有相互作用。
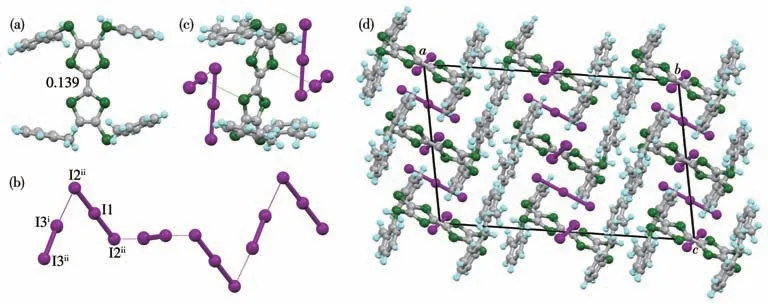
图3 复合物(1+·)(I3)·I2的晶体结构:(a)1+·的俯视图及其中心C=C键长(nm);(b)I2和I3-的构型;(c)1+·与I3-和I2之间的分子间相互作用;(d)晶体堆积结构Fig.3 Crystal structure of(1+·)(I3)·I2:(a)top view of 1+·and the central C=C bond lengths(nm);(b)anion sheets composed of I3-and I2;(c)interactions between 1+·with I3-and I2;(d)packing structure
2.4 复合物(2+·)(I5)·I2的晶体结构
复合物(2+·)(I5)·I2为黑色块状晶体,属于三斜晶系空间群。不对称单元中,包括一个2+·、一个I5-阴离子和一个I2。如图4 所示,2+·为椅式构型,中心C=C 键长为0.138 nm,理论计算δ为0.071 7,根据Day 等的经验公式,化合物2 在复合物中显+1 价。沿b轴方向,2+·呈柱状堆积,分子间以S…Cl(0.345~0.352 nm)相互作用;沿a轴方向,2+·之间以苯环碳原子与外围硫原子间的S…C(0.346~0.347 nm)相互作用;复合物中2+·呈二维网格状堆积。I1与I2通过共价键形成中性的I2,I1—I2 键长为0.273 nm。I3—I4—I5—I6—I7 形成“V”字形的I5-离子,夹角为81.66°。I3—I4(0.281 nm)和I6—I7(0.275 nm)的键长在I2(0.273 nm)和I3-(0.290 nm)键长之间,I4—I5(0.301 nm)和I5—I6(0.317 nm)的键长在碘原子形成高聚物键长的范围[27-28]。I5-和I2通过I…I(0.339~0.394 nm)的范德瓦耳斯力相互连接,形成二维网格状,如图4c所示。2+·垂直插入碘原子形成的二维网格,通过I…S(0.366~0.378 nm)和I…C(0.367~0.353 nm)与碘形成紧密作用,呈三维网格状堆积。
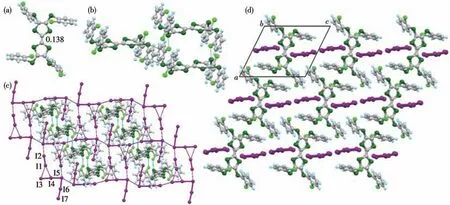
图4 复合物(2+·)(I5)·I2的晶体结构:(a)2+·的俯视图及其中心C=C键长(nm);(b)2+·分子间作用;(c)碘阴离子的二维网格状结构及其与2+·的位置关系;(d)晶体堆积结构Fig.4 Crystal structure of(2+·)(I5)·I2:(a)top view of 2+·and the central C=C bond lengths(nm);(b)interactions between 2+·;(c)2D grid structure of iodide anions and its relation to the position of 2+·;(d)packing structure
2.5 复合物(32+)(I3)2的晶体结构
复合物(32+)(I3)2为黑色片状晶体,属于正交晶系Pbca空间群。不对称单元包括一个32+和2 个I3-阴离子。如图5 所示,复合物中32+外围4 个吡啶环与中心骨架基本处于同一平面,这与已经报道的Ar-STTF 衍生物中性状态和氧化状态的分子构型均不同[21,24-25,30-34]。32+的中心C=C 键长为0.143 nm,理论δ为0.061 5,根据Day 等的经验公式,化合物3 在复合物中显+2价。复合物中化合物3比溶液中呈现更高的氧化态,这与化合物3 的氧化还原性和复合物堆积结构有密切关系。复合物中32+外围硫原子与芳香环上碳原子之间通过S…C(0.349 nm)与相邻的32+相互作用,沿a轴方向成柱状堆积,如图5b 所示;通过芳香环上碳原子和氢原子之间的2 条C…H(0.285 和0.287 nm)与相邻非平行的32+相互作用。复合物中32+独特的平面分子构型,进一步证明了Ar-S-TTF的芳基自由旋转性。
图5 复合物(32+)(I3)2的晶体结构:(a)32+的俯视图、中心C=C键长及其与碘阴离子的相互作用(nm);(b)32+间相互作用;(c)晶体堆积结构Fig.5 Crystal structure of(32+)(I3)2:(a)top view of 32+,the central C=C bond lengths and interactions between 32+and I3-(nm);(b)interactions between 32+;(c)packing structure
I1—I2—I3 和I4—I5—I6 的I—I 键长为0.287~0.298 nm,这与I3-的键长0.290 nm 一致,说明I1—I2—I3 和I4—I5—I6 均为I3-[27-29]。I1—I2—I3 和I4—I5—I6通过I…I(0.367 nm)相互作用,形成一维链状,这也说明在复合物中化合物3呈+2价。I3-通过I…S(0.362 和0.371 nm)和I…H(0.296 和0.302 nm)与相邻的32+相互作用,形成碘和化合物3 的电荷转移复合物。
3 结 论
采用Ar-S-TTF化合物1~3作为给电子化合物与碘反应,采用缓慢挥发溶剂的方法合成了3 种电荷转移复合物:(1+·)(I3)·I2、(2+·)(I5)·I2和(32+)(I3)2。化合物2 和3 在电荷转移复合物中可观察到比在碘的溶液中呈现更高的氧化态,这与复合物中电子给体和电子受体紧密的分子堆积有关。Ar-S-TTF 作为电子给体能够通过改变自身取代基调控客体分子构型,使碘聚物呈现不同的一维和二维聚合结构。另一方面,复合物中化合物1~3 可依据客体分子结构调整自身分子构型,化合物1 和2 呈不同的椅式构型,而化合物3 呈独特的平面构型。结合之前的报道,本工作进一步证明了Ar-S-TTF 独特的性质,这是由于其电子态和分子几何结构可依据客体分子而改变。这对合成具有独特性质的电荷转移复合物具有重要意义。
