幽门螺旋杆菌尿素酶纳米抗体UreNb重组质粒的构建及其产物的表达、纯化与鉴定※
2023-10-12刘文婧吕慧玲王顺娟李润乐
刘文婧,吕慧玲,王顺娟,冯 琳,汤 锋,李润乐§,胥 瑾*
(1.青海大学医学部,西宁 810001;2.青海省藏医院,西宁 810007;3.青海省人民医院,西宁 810007;4.青海大学高原医学研究中心,西宁 810001)
课题组前期利用羊驼天然纳米抗体文库,筛选得到了针对幽门螺旋杆菌(Helicobacterpylori,H.pylori)尿素酶B亚基(Urease-B)的高亲和力纳米抗体UreNb。本研究构建幽门螺旋杆菌尿素酶纳米抗体UreNb重组质粒,在大肠杆菌系统中通过诱导表达目的蛋白并予以纯化,再对表达产物及纯化产物进行鉴定。
1 材料与方法
1.1 材料
1.1.1 主要试剂
pET22b质粒、DH5α感受态细胞、Arctic-Express表达菌、BL-21(DE3)表达菌、Rosetta表达菌、pSumo-mut质粒均购自钟鼎生物公司。限制性内切酶购自TaKaRa公司,蛋白质Marker购自Thermo公司,IPTG购自Biotopped公司,氨苄青霉素钠、四环素、卡纳霉素购自Solarbio公司,0.22 μm无菌滤器和透析袋购自Ruitaibi公司,Ni-IDA亲和层析柱购自GenScript公司,4×蛋白上样缓冲液购自Solarbio公司。
1.1.2 主要仪器
多聚酶链式反应仪购自艾本德公司,凝胶成像分析系统购自法玛西亚公司,酶标分析仪购自美谷分子公司,蛋白纯化仪购自上海嘉鹏有限公司,低温高速离心机购自德国西格玛公司。
1.2 实验方法
1.2.1 UreNb在质粒pET22b中的构建及表达鉴定
在前期研究中经筛选获得了针对Urease-B纳米抗体UreNb的氨基酸序列,经翻译后得到的核苷酸序列采用密码子优化后用于全基因合成。在T1连接酶体系下经过酶切(NcoI/XhoI)后连接(4℃,过夜),将UreNb核苷酸序列克隆至质粒pET22b后转化入大肠杆菌DH5α感受态细胞,获得重组质粒pET22b-UreNb。从培养的阳性菌落挑选、提取质粒,通过双酶切(MluI/XhoI)法鉴定克隆结果。pET22b-UreNb分别转化至表达菌株BL-21、Arctic Express、Rosetta,涂布于含有相应抗性的固体培养基上培养(37℃,过夜)后,挑取单克隆并进行扩大培养,而后分别吸取1 mL菌液加入到200 mL的LB培养基中扩增培养。三种表达菌同时加入异丙基-β-D-硫代半乳糖苷(Isopropyl-β-D-thiogalactoside,IPTG。10 mmo/L)进行诱导。通过优化诱导表达条件,使用BL-21表达菌株时,将诱导温度分别调整至15℃和37℃,使用Arctic Express、Rosetta菌株表达时,将诱导温度调整至37℃。随后分别从三个表达菌株中取出1 mL菌液离心后弃上清,将1×上样缓冲液(100 μL)加入沉淀物中重悬。剩余培养物离心后弃上清用PBS重悬菌体沉淀物,将重悬液用超声波破碎后,分别取上清与沉淀物加入上样缓冲液后重悬。做电泳(12% SDS-PAGE)分析后以考马斯亮蓝染色确认诱导表达结果。
1.2.2 UreNb在质粒pSumo-mut中的构建及表达鉴定
用T1连接酶体系连接,将经UreNb纳米抗体经过密码子优化后的核苷酸序列克隆至pSumo-mut表达载体,克隆位点为无缝克隆-XhoI。电转至大肠杆菌DH5α感受态细胞,挑选阳性单克隆菌落并提取质粒,通过酶切(MluI/XhoI)鉴定质粒pSumo-mut-UreNb的构建结果。随后将正确克隆的重组质粒转化至BL-21,将阳性克隆菌落接种于Kana LB液体培养基(5 mL)中(37℃,220 r/min,过夜),培养后按1%接种于Kana LB液体培养基(50 mL)中培养(37℃,220 r/min)至OD值为0.4~0.6,加入IPTG诱导(15℃,过夜)。次日收集菌体,用超声波破碎后收集上清及沉淀物。将诱导前、后的菌液以及超声波破碎后的上清和沉淀物进行SDS-PAGE电泳,以确认诱导表达结果。
1.2.3 重组蛋白pSumo-mut-UreNb的包涵体复性
对上述表达产物鉴定,重组蛋白pSumo-mut-UreNb在沉淀物中表达,遂将上一步骤中经超声波破碎后的菌液重悬、离心后弃上清。沉淀物分别用50 mM NaH2PO4、10 mM Tris-HCL、8 M尿素溶液混悬后以超声波裂解。将上述溶液离心后吸取上清加入透析袋中,先后置于添加了还原型谷胱甘肽和氧化型谷胱甘肽的复性工作缓冲液中,静置(4℃,过夜)后离心收集上清。
1.2.4 pSumo-mut-UreNb融合蛋白的纯化分析
使用经Binding-Buffer(0.15 M NaCl,20 mM Tris-HCl,pH为8.0)预平衡的亲和层析柱,冲洗至流出液OD280值到达基线后加入复性后的蛋白溶液。用Washing-Buffer(20 mM Tris-HCl,20 mM Imidazole,0.15 M NaCl,pH为8.0)冲洗,至流出液OD280值到达基线。用Elution-Buffer(20 mM Tris-HCl,250 mM Imidazole,0.15 M NaCl,pH为8.0)洗脱目的蛋白,收集流出液进行SDS-PAGE分析。
1.2.5 pSumo-mut-UreNb纯化蛋白鉴定
将1.2.4步骤中的凝胶用湿转法将蛋白转移至PVDF膜上,以鼠源His-tag为一抗、HPR标记羊抗鼠IgG为二抗,添加显色液使用荧光化学发光仪成像进行Western Blot验证。
2 结果
2.1 pET22b-UreNb的质粒构建及表达鉴定结果
Ure-Nb目的基因经密码子优化后得到的核苷酸序列如下:CCATGGGCCAGGTTCAGCTGGTGGAAAGCGGCGGTGGTCTGGTTCAGGCAGGTGGTTCACTGCGCCTGAGCTGTGCTGCTAGCGGTAATACCCTGCGTATTGTTGCAATGGGTTGGTATCGCCAGGGTGCAGGCAATAAACGTGATCTGGTGGCAAGTATTAGCACCAGCAATGATCGTACCACCTATGCCGATAGCGTGAAAGGCCGCTTTACCATTAGTCGTGATAGTGCAAAAAACACCATGTATCTGCAGATGAATAGCCATCATCATCATCACCATTGACTCGAG。目的序列连入载体pET22b后经酶切(MluI/XhoI)的鉴定结果见图1。图1显示,克隆位点与酶切位点不在同一位置,图中条带显示的片段大小与预期设计大小一致(1 200 bp),表明纳米抗体UreNb目的序列已成功插入质粒中,表示重组表达载体pET22b-UreNb构建成功。
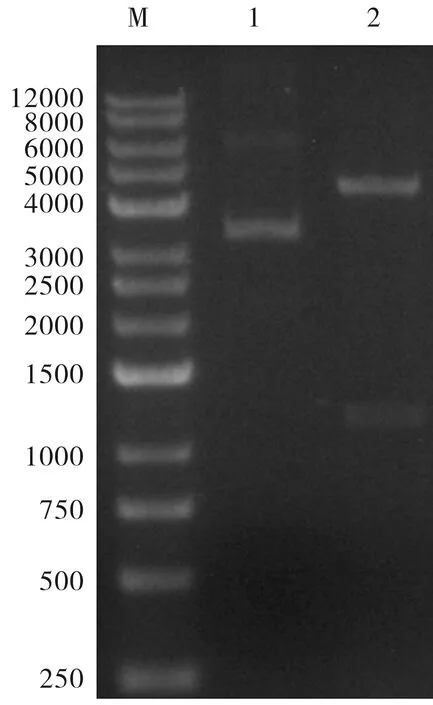
M:Marker;Line1:酶切前质粒;Line2:酶切后质粒
将pET22b-UreNb分别转化至表达菌株BL-21、Arctic Express、Rosetta中,其中图2、3显示BL-21表达菌株在诱导温度为15、37℃时未出现表达条带。
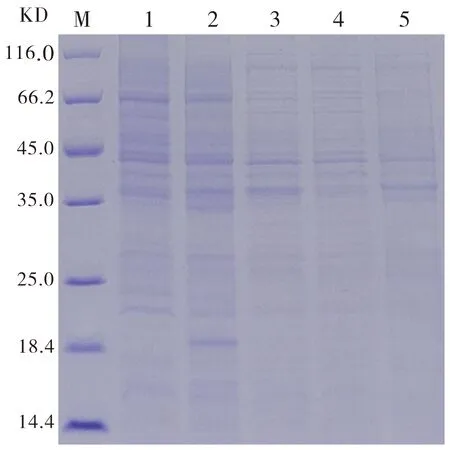
M:Marker;Line1:pET22b诱导(空载);Line2:未诱导;Line3:诱导后;Line4:诱导破碎后上清;Line5:诱导破碎后沉淀物
M:Marker;Line1:pET22b诱导(空载);Line2:未诱导;Line3:诱导后;Line4:诱导破碎后上清;Line5:诱导破碎后沉淀物
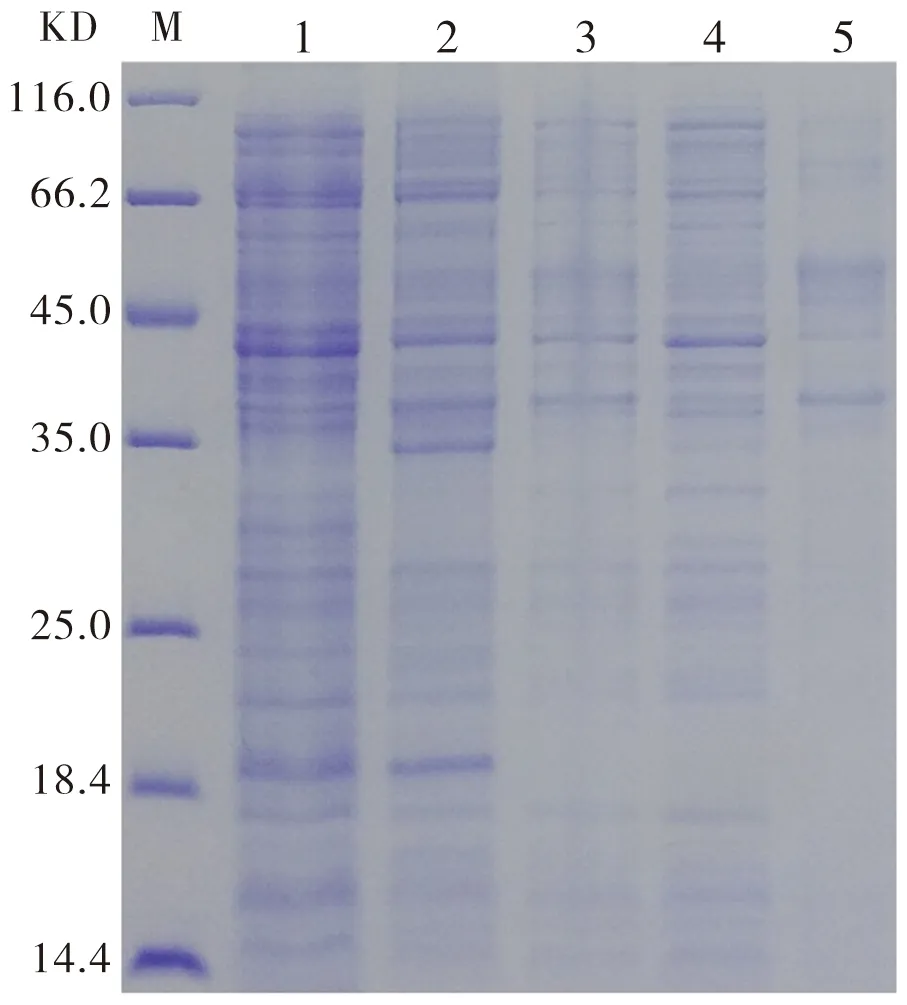
图4、5显示表达菌Arctic Express、Rosetta被诱导(37℃)后未见明显表达,故更换重组表达载体为pSumo-mut。

M:Marker;Line1:pET22b诱导(空载);Line2:未诱导;Line3:诱导后;Line4:诱导破碎后上清;Line5:诱导破碎后沉淀物
M:Marker;Line1:pET22b诱导(空载);Line2:未诱导;Line3:诱导后;Line4:诱导破碎后上清;Line5:诱导破碎后沉淀物
2.2 pSumo-mut-UreNb的质粒构建及表达鉴定结果
经T1连接酶体系连接,将UreNb插入原核表达载体pSumo-mut,克隆位点为无缝克隆-XhoI,序列如下:CAGGTTCAGCTGGTGGAAAGCGGCGGTGGTCTGGTTCAGGCAGGTGGTTCACTGCGCCTGAGCTGTGCTGCTAGCGGTAATACCCTGCGTATTGTTGCAATGGGTTGGTATCGCCAGGGTGCAGGCAATAAACGTGATCTGGTGGCAAGTATTAGCACCAGCAATGATCGTACCACCTATGCCGATAGCGTGAAAGGCCGCTTTACCATTAGTCGTGATAGTGCAAAAAACACCATGTATCTGCAGATGAATA GCTAACTCGAG。目的序列连入载体pSumo-mut后,提取质粒经酶切(MluI/XhoI)鉴定,条带大小与预期大小一致(1 500 bp),表明UreNb目的序列已成功插入质粒中,表示重组表达载体构建成功,见图6。
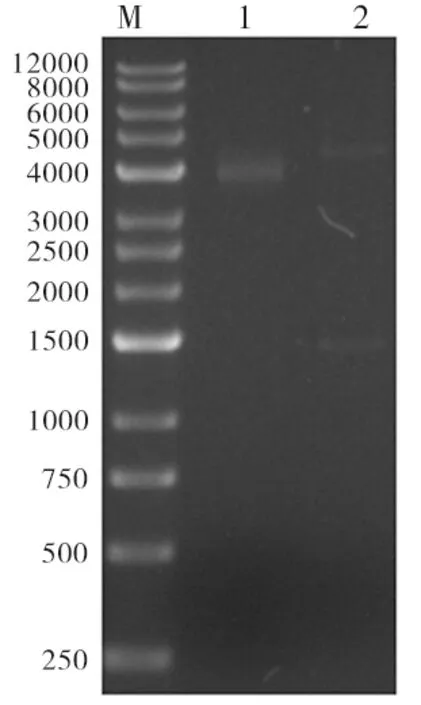
M:Marker;Line1:酶切前质粒;Line2;酶切后质粒
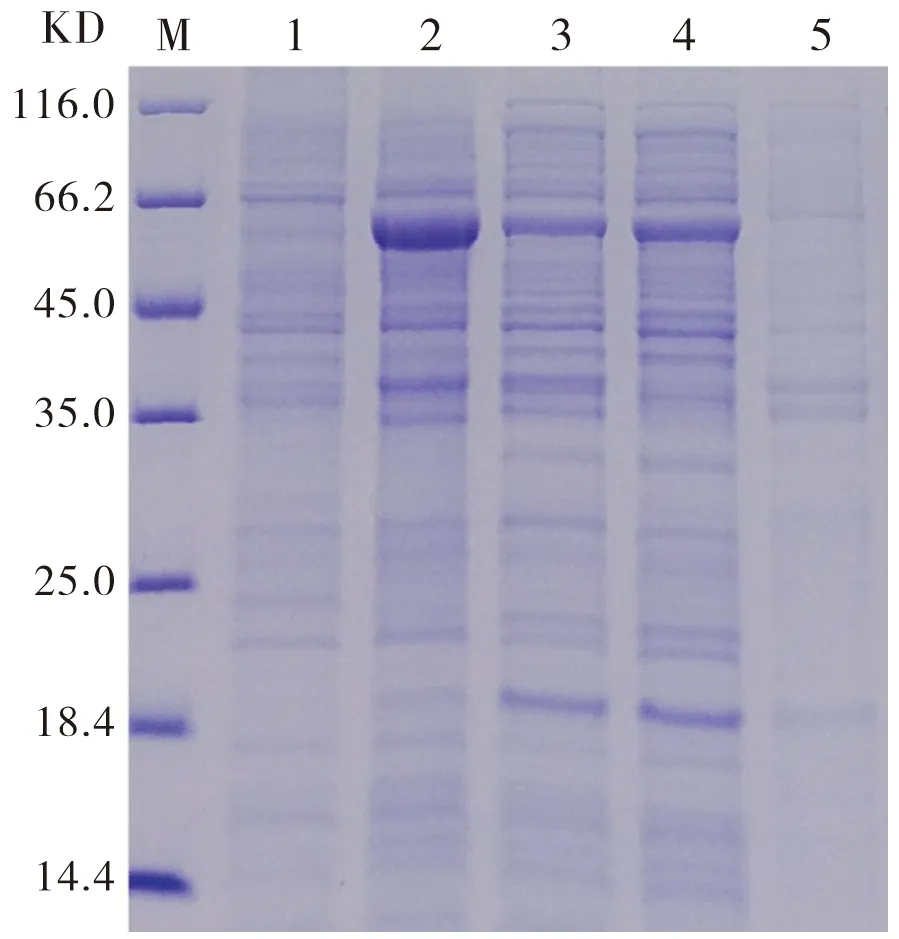
经IPTG诱导表达蛋白pSumo-mut-UreNb,对未诱导样本、诱导后样本及破碎后上清和沉淀进行SDS-PAGE分析。图7显示,在破碎后沉淀部位27 kD处可见明显增粗条带,而在可溶条带中未见。上述实验结果证明,原核表达载体BL-21能够成功表达重组蛋白pSumo-mut-UreNb,且重组蛋白pSumo-mut-UreNb在原核表达载体BL-21中是以包涵体形式表达。
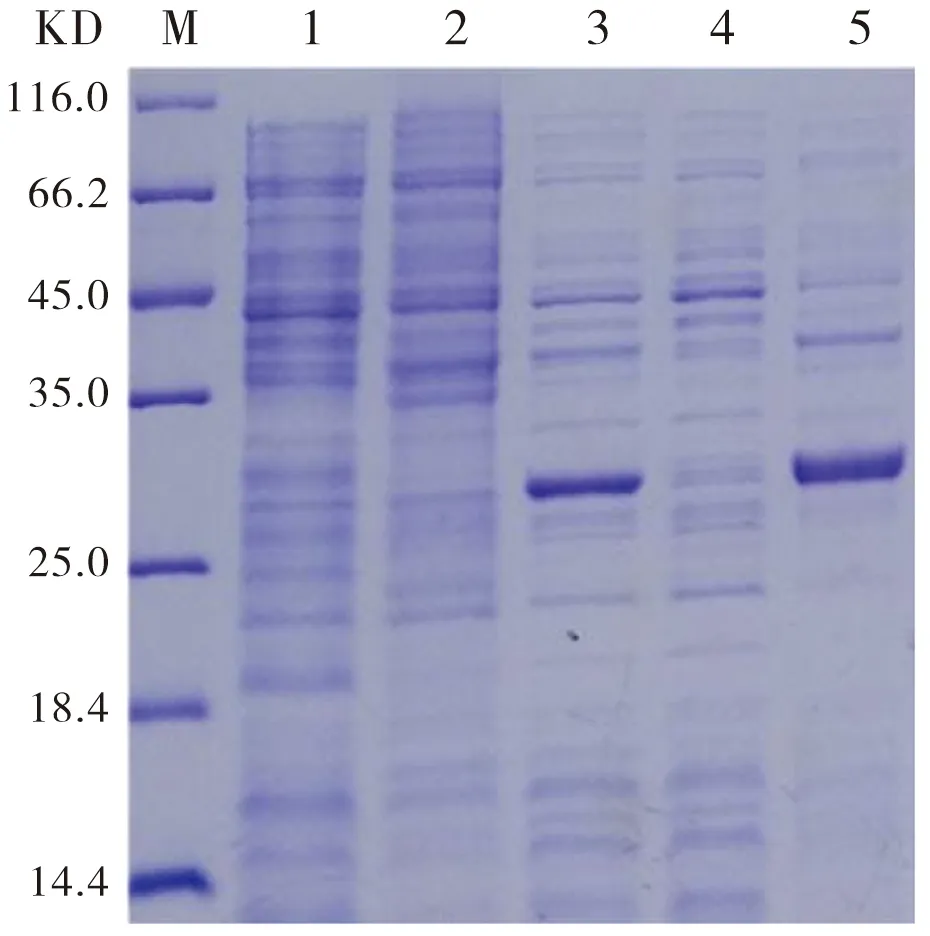
M:Marker;Line1:pET22b诱导(空载);Line2:未诱导;Line3:诱导后;Line4:诱导破碎后上清;Line5:诱导破碎后沉淀物
2.3 重组蛋白pSumo-mut-UreNb的包涵体复性和纯化结果
目标蛋白经包涵体变、复性和纯化后对超声波破碎处理后的样品、纯化后流出样品及洗脱样品进行SDS-PAGE分析。图8显示,纯化后的目的蛋白纯度较高。
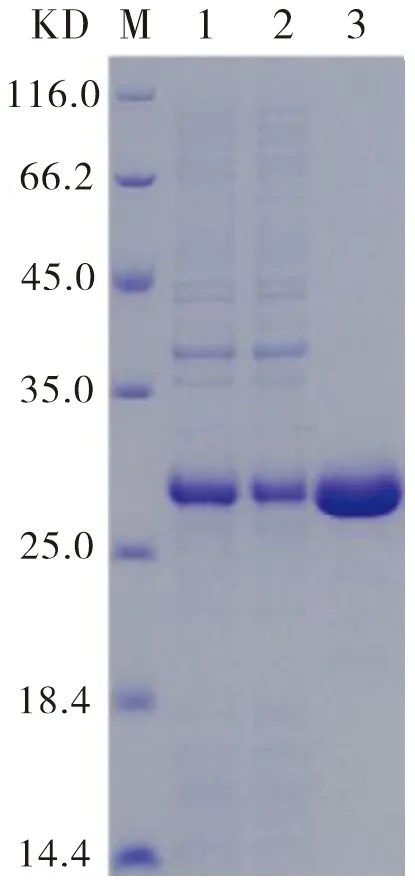
M:Marker;Line1:未纯化蛋白;Line2:洗脱液中的蛋白;Line3:纯化后的目的蛋白
对纯化后获得的目的蛋白以鼠源His-tag为一抗、HPR标记羊抗鼠IgG为二抗进行Western Blot验证,结果显示,目的蛋白大小约为27 kDa左右,与理论分子量基本吻合,结果符合实验预期结果,见图9。
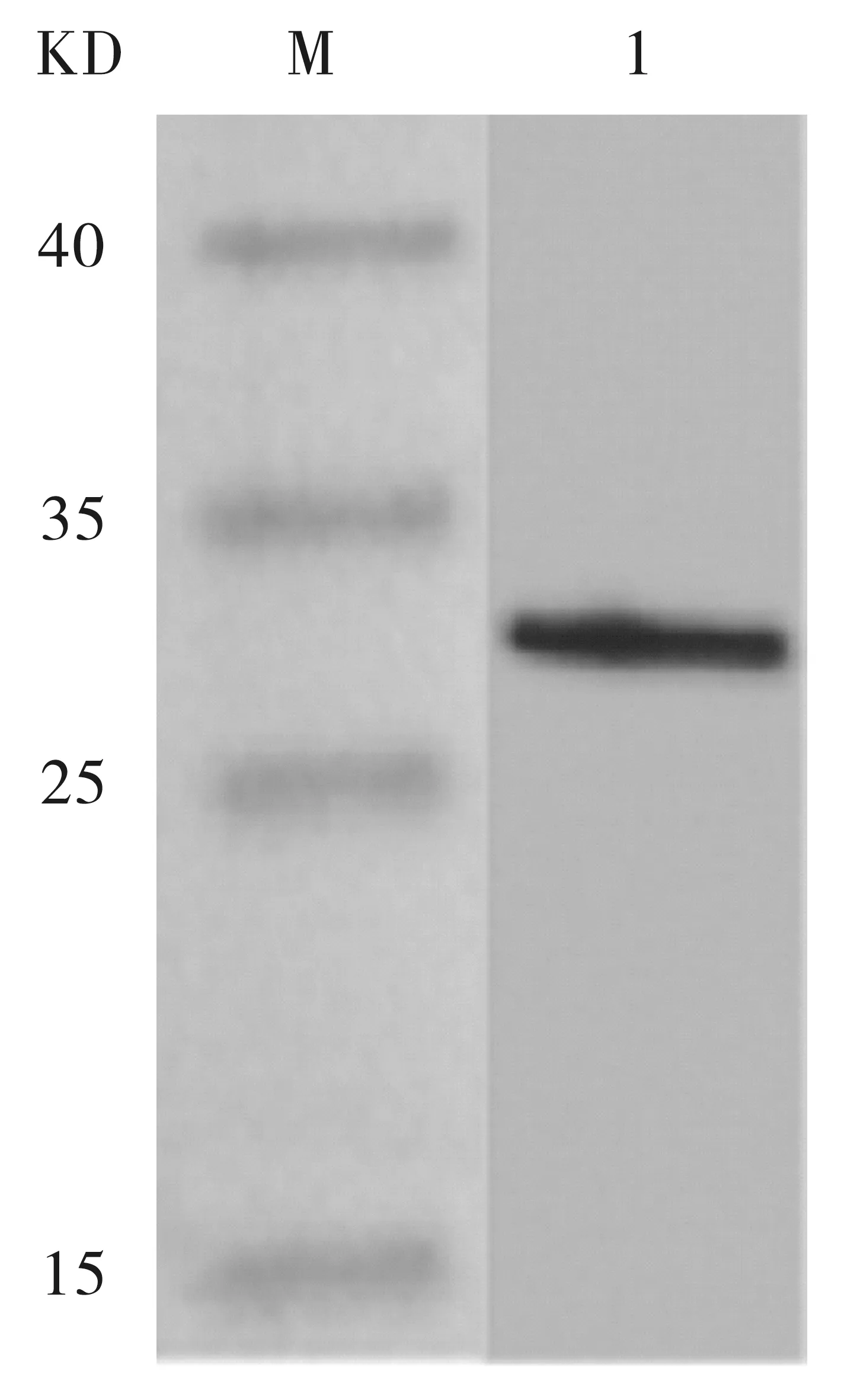
M:Marker;1:纯化后样品
3 讨论
目前,单克隆抗体应用于检测及诊断治疗方面的研究较多,但小分子纳米抗体的应用还在起步阶段[1],尤其在幽门螺旋杆菌感染治疗方面还未出现用于诊断及治疗的纳米抗体。幽门螺旋杆菌作为胃部特异性病原菌,是导致胃部疾病发生发展的重要细菌,其中Urease的表达贯穿于幽门螺旋杆菌整个生命活动的始终[2]。本研究中尿素酶纳米抗体能够特异性结合Urease-B亚基,使其失去活性。另一方面,Urease-B含有酶活性中心,具有很高的抗原性,是开发预防和治疗性疫苗的潜在抗原[3]。纳米抗体天然存在于骆驼科及鲨鱼科动物血清中,与传统单克隆抗体不同,纳米抗体的独特结构和小分子量,以及高稳定性为抗体药物的开发提供了全新的思路,同时也可因需要而进行人源化改造[4]。因此寻找合适的表达载体,并且能够高效、大量表达此抗体,可有助于后期的实验研究。
本实验在抗体表达过程中首先选择了pET-22b质粒,此质粒能够将表达的目的蛋白定位在细胞外周质腔后形成可溶性蛋白[5]。随后使用表达载体pSumo-mut,此质粒不仅能够提高蛋白的溶解性,而且有助于诱导蛋白形成正确折叠,从而形成三级空间结构[6]。其携带的6×His标签使重组蛋白可以通过Ni柱亲和纯化后获得高纯度的蛋白。利用Sumo蛋白酶能够特异性切割融合蛋白,从固定的位置将pSumo-mut蛋白与重组蛋白彻底分离[7]。在原核表达系统中,BL-21大肠杆菌表达菌株不含lon、OmpT蛋白酶,所以可降低细胞中表达的异源蛋白的降解水平并且提高转化效率[8]。ArcticExpress菌株是BL-21的改良菌株,可促进表达蛋白的稳定,此表达菌可降低重组蛋白包涵体的形成率,增加可溶重组蛋白的表达量及生物活性[9]。Rosetta菌株可补充大肠杆菌缺乏的6种稀有密码子对应的tRNA,提高外源基因的表达水平[10]。本研究首先选择pET-22b载体拟提高可溶蛋白表达率,然后选择Arctic Express、BL-21、Rosetta三种表达菌分别测试重组蛋白表达成功率,结果显示,成功构建的pET-22b-UreNb均未在上述三种表达菌中有效表达。随后将重新构建的重组质粒pSumo-mut-UreNb转化入表达菌BL-21中进行表达,经鉴定可在沉淀物中大量表达,纯化后经过SDS-PAGE、Western Blot验证,鉴定其为成功表达载体,成功获得UreNb纯化蛋白。pSumo-mut载体因携带Sumo融合蛋白,能够使目的蛋白形成正确折叠,且后续经特异性切割融合蛋白后,重组蛋白N端无氨基残留,有利于后续研究的进行。
青海地区幽门螺旋杆菌感染的发生率较高,其感染与胃溃疡、胃炎以及胃癌的发生也有一定相关性[11]。幽门螺旋杆菌感染存在于疾病的活动期和静止期,活动期感染增殖速度更加迅速,患者临床症状更为明显[12]。在幽门螺旋杆菌的刺激下,不但可以诱发胃癌,而且能够影响胃癌的浸润及转移。因此,在高原地区胃部疾病的防治中,控制幽门螺旋杆菌感染是现阶段仍需解决的重要问题。
鉴于以上纳米抗体UreNb的应用前景,本研究基于前期研究基础,构建了该纳米抗体的重组质粒,建立了原核表达体系,经纯化得到了其纯化蛋白,为制备幽门螺旋杆菌诊断抗体开辟了新的途径。
