青海成年痰检阳性与阴性活动性肺结核患者的肠道菌群差异性※
2023-10-12王玉清王兆芬程国栋史发鹏冶庚祉黄秋丽曹雪平
王 玲,王玉清,王兆芬#,姚 杰,程国栋,史发鹏,冶庚祉,黄秋丽,曹雪平,李 泽
(1.青海大学医学院,西宁 810001;2.青海省第四人民医院,西宁 810007)
对肺结核患者进行精准化治疗需要明确痰检阳性与阴性活动性肺结核患者的肠道菌群差异情况。为了探究青海成年痰检阳性与阴性活动性肺结核患者的肠道菌群差异性,本研究利用16S rDNA测序技术开展相关研究。
1 对象与方法
1.1 对象
本研究依据《肺结核诊断标准(WS 288-2017)》,选取青海省第四人民医院痰检阳性活动性肺结核患者14例作阳性组、痰检阴性活动性肺结核患者12例作阴性组。研究对象年龄范围为18-85岁。痰检阳性组与阴性组肺结核患者基本情况相似。研究方案通过青海大学医学院伦理委员会审核。
1.2 方法
1.2.1 粪便采集
在抗结核治疗前采集3~10 g粪便置-80℃冰箱保存。使用QIAamp PowerFecal DNA Pro Isolation Kit试剂盒提取细菌基因组DNA,经琼脂糖凝胶电泳确定基因组DNA完整性后,使用Nanodrop 2000平台(Thermo Fisher Scientific,美国)检测基因组DNA质量(浓度为20 ng/μL,总量≥500 ng,OD260/280=1.8~2.0)。
1.2.2 16S rDNA基因测序
使用PCR扩增引物对细菌总DNA中的16S rDNA基因的V3-V4区进行扩增。16S rDNA V3-V4区的扩增引物序列见表1。按建库试剂盒说明(Illumina Inc,美国)在扩增产物的两端加上barcode序列,等比例混合该产物后,由上海天昊生物有限公司在Illumina NovaSeq 6000平台上测序。
1.2.3 生物信息学分析
使用QIIME2软件的DADA2插件对数据进行质量过滤、降噪、拼接及去嵌合体处理。使用QIIME2软件对ASV/OTU(Amplicon sequence variant,ASV/Operational taxonomic units,OTU)序列进行分类学注释,并计算α多样性指数(Chao1、ACE、Shannon及Simpson指数)和β多样性指数。
1.2.4 统计学分析
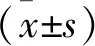
2 结果
2.1 肠道菌群差异
2.1.1 样本测序量
26份样本经16S rDNA测序后,共得到2 449 062条原始序列,对其双端序列进行质量控制、拼接及去噪后共获得1 771 789条高质量序列,共生成2 341个ASV,其中痰检阳性组与痰检阴性组共有597个ASV,痰检阴性组特有的ASV有775个,痰检阳性组特有的ASV有969个,见表2。

表2 痰检阳性组和痰检阴性组样本的ASV数量
2.1.2 肠道菌群多样性与丰富度
α多样性分析显示,成年痰检阴性肺结核患者与痰检阳性肺结核患者的肠道菌群的Chao1指数、ACE指数、Shannon指数及Simpson指数无明显统计学差异(P>0.05),见表3。β多样性分析显示,两组样本肠道菌群的多样性差异无统计学意义,见图1。经PERMANOVA检验显示,两组差异没有统计学意义(P>0.05)。
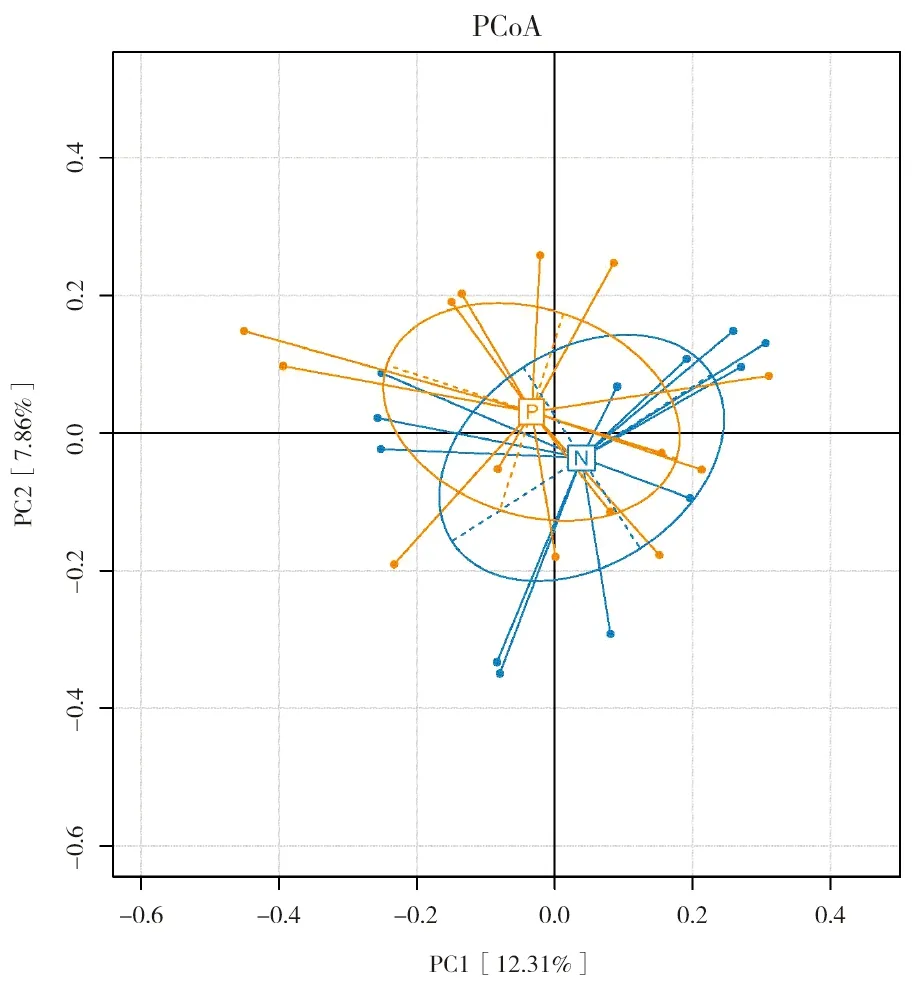
P:痰检阳性组;N:痰检阴性组

表3 痰检阳性组和痰检阴性组样本α多样性指数
2.2 门和属水平上的物种组成和丰度差异
利用Silva数据库(http://www.arb-silva.de)对获得的代表性序列ASV进行物种注释,采用R软件包分别从门水平和属水平对两组研究对象的菌群相对丰度值进行分析。26份样本共检测出11个门、18个纲、26个目、56个科、140个属。在门级别中,两组研究对象中相对丰度含量较高的菌门是厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)和疣微菌门(Verrucomicrobia)等,见表4。使用Metastats方法进行差异比较,痰检阴性组与阳性组相比,厚壁菌门相对丰度差异无统计学意义(57.06% vs 47.49%,P>0.05),拟杆菌门相对丰度差异无统计学意义(32.10% vs 26.86%,P>0.05),见表5。在属水平中,两组研究对象中相对丰度含量较高的菌属分别是拟杆菌属(Bacteroides)、普雷沃氏菌属(Prevotella)、粪杆菌属(Faecalibacterium)、双歧杆菌属(Bifidobacterium)和埃希氏志贺氏菌属(Escherichia/Shigella),见表4。痰检阴性组与阳性组相比,拟杆菌属相对丰度值(19.02% vs 13.47%,P>0.05)、普雷沃氏菌属相对丰度值(6.69% vs 6.5%,P>0.05)和粪杆菌属相对丰度值(8.37% vs 4.46%,P>0.05)的差异均无统计学意义(P>0.05),见图2。在门和属水平上,痰检阳性组与阴性组的肠道菌群丰度差异存在统计学意义,见表6。

表4 总体样本肠道菌群丰度前5的物种相对丰度值
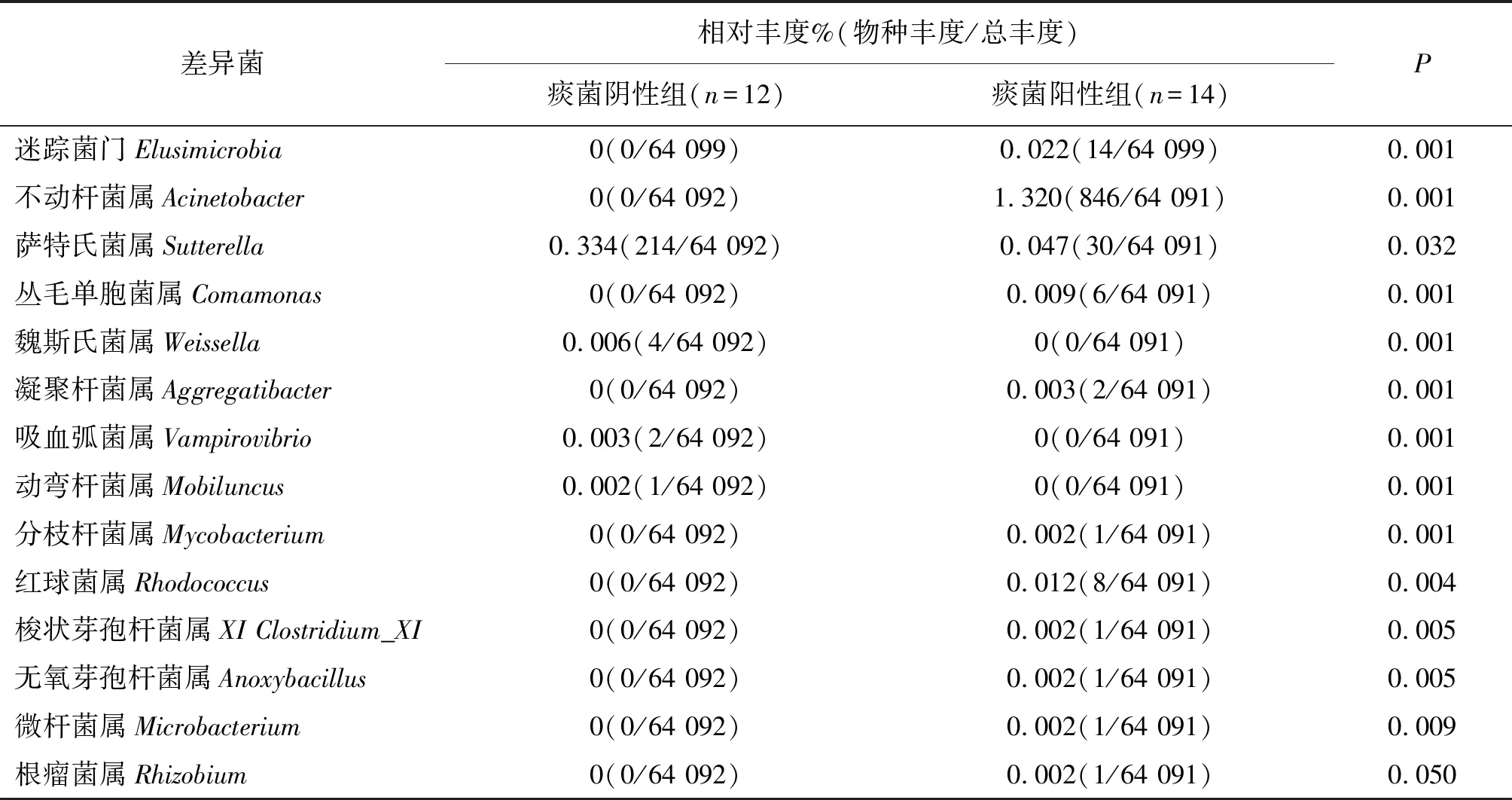
表6 痰检阴性与痰检阳性组门和属水平中肠道菌的相对丰度
2.3 线性判别分析结果
经LEfSe分析,进一步筛选出2个最可能解释阴性组与阳性组差异的肠道菌群,分别是红球菌属和诺卡氏菌科。痰检阴性组与阳性组的|LDA|值在红球菌属(0 vs 2.530)和诺卡氏菌科(0 vs 2.431)中的差异均具有统计学意义(P<0.05)。
3 讨论
3.1 肠道菌群差异
本研究发现,所调查的青海成年痰检阳性肺结核患者与痰检阴性患者的肠道菌群多样性和丰富度差异在统计学上无意义。研究显示[1],肺结核感染与肠道菌群多样性无关联。研究显示[2],经过一周和一个月治疗的肺结核患者的肠道菌群多样性与丰富度无明显差异。
3.2 门和属水平上的物种组成和丰度差异
肠道菌群构成分析显示,两组研究对象的肠道菌群中厚壁菌门、拟杆菌门与变形菌门为三组优势菌门,共占总菌门数80%以上。痰检阴性组与阳性组相比,厚壁菌门相对丰度和拟杆菌门相对丰度的差异无统计学意义。最近的研究结果表明,短链脂肪酸(short-chain fatty acids,SCFAs)中的丁酸盐具有重要的肠道和免疫调节功能[3],而丁酸盐主要由厚壁菌门产生。SCFAs通过不同的机制调节肠上皮细胞的增殖和分化,加强肠道屏障功能以及宿主代谢。SCFAs通过强化NF-κB的活化来影响促炎细胞因子(例如IL-6和TNFα)的产生[4]。在炎症期间,SCFAs通过激活G蛋白偶联受体维持免疫稳态[5]。SCFAs在紧密连接蛋白的协同下维持肠上皮屏障的完整性[6]。同时,特异性SCFAs通过Keap1-Nrf2途径防止氧化损伤[7]。两组间差异菌门为迷踪菌门,在痰检阳性组相对丰度较高。两组研究对象共鉴定出13种差异菌属,其中痰检阳性组中不动杆菌属、丛毛单胞菌属、凝聚杆菌属、分支杆菌属、红球菌属、梭菌属、无氧芽孢杆菌属、微杆菌属及根瘤菌属的相对丰度较高。研究显示[8],肠道菌群中不动杆菌属是条件致病菌,不动杆菌的毒力特性主要来源于先天免疫系统对其清除的逃避,导致不动杆菌数量增加,从而引发脂多糖(Lipopolysaccharide,LPS)——TLR4介导的败血症。不动杆菌属在痰检阳性组富集,提示痰检阳性患者可能易受到细菌攻击。目前,已有研究将菌群失调与感染结核分枝杆菌后的免疫损伤联系起来,发现微生态平衡破坏导致结核病易感性增加。研究显示[9],小鼠在感染结核分枝杆菌的第一周出现微生物群失调,导致结核分枝杆菌在肺部的早期定植增加,这与此段时间内肠道微生物群的多样性改变有关;研究显示[10],用抗结核药异烟肼和吡嗪酰胺治疗小鼠导致的肠道微生物组变化与肺结核传播加快有关。
3.3 线性判别
恰当的生物标志物可以辅助肺结核的鉴定和治疗。本研究发现,红球菌属和诺卡氏菌科可作为区分青海成年痰检阴性活动性肺结核患者与痰检阳性活动性肺结核患者的潜在差异标志物。但因样本量少,需进一步确认。
综上所述,青海成年痰检阳性与阴性活动性肺结核患者的肠道菌群的多样性和丰富度无差异性,但菌群构成差异显著,特定红球菌属与诺卡氏菌科菌群可作为潜在生物标志物。
