缺血性脑卒中的线粒体质量控制与铁死亡关联机制及中医药干预研究进展
2023-09-07韦梦真,刘丹红,曾劲松,方锐,梅志刚,葛金文,廖君
韦梦真,刘丹红,曾劲松,方锐,梅志刚,葛金文,廖君


〔摘要〕 缺血性脑卒中(ischemic stroke, IS)是一个复杂的级联过程,对大脑产生不可逆的损伤,严重危害人类健康。铁死亡是铁依赖及脂质过氧化物生成所致的一种细胞死亡方式,是IS的一个重要病理表现。线粒体是真核生物进行氧化代谢的细胞器,是糖类、脂肪和氨基酸最终氧化释放能量的场所。线粒体质量控制(mitochondrial quality control, MQC)是一个内源性调控机制,作为氧化代谢的主要调节器和铁利用的主要场所,MQC与铁死亡密切关联。稳定MQC是抑制铁死亡保护IS的重要治疗机制。系统介绍在IS中MQC与铁死亡关联机制,为IS发生机制研究和临床药物靶点提供新思路。
〔关键词〕 缺血性脑卒中;线粒体质量控制;线粒体自噬;线粒体动力学;线粒体生物合成;铁死亡;中医药干预
〔中图分类号〕R255 〔文献标志码〕A 〔文章编号〕doi:10.3969/j.issn.1674-070X.2023.08.028
Research progress on the correlation mechanism between mitochondrial quality control and ferroptosis in ischemic stroke and the TCM intervention
WEI Mengzhen, LIU Danhong, ZENG Jinsong, FANG Rui, MEI Zhigang, GE Jinwen, LIAO Jun*
Hunan University of Chinese Medicine, Changsha, Hunan 410208, China
〔Abstract〕 Ischemic stroke (IS) is a complex cascade process, which causes irreversible damage to the brain and seriously endangers human health. Ferroptosis is a form of cell death caused by iron-dependence and lipid peroxide generation, and is an important pathological manifestation of IS. Mitochondria are eukaryotic organelles for oxidative metabolism, and are the sites where sugars, fats and amino acids eventually oxidize and release energy. Mitochondrial quality control (MQC) is an endogenous regulatory mechanism. As the main regulators of oxidative metabolism, mitochondria are also the main sites of iron utilization, so MQC is closely correlated with ferroptosis. Stabilizing MQC is an important therapeutic mechanism for inhibiting ferroptosis and protecting the body from IS. This article systematically introduces the correlation mechanism between MQC and ferroptosis in IS to provide new ideas for the study of IS pathogenesis and clinical drug targets.
〔Keywords〕 ischemic stroke; mitochondrial quality control; mitophagy; mitochondrial dynamics; mitochondrial biogenesis; ferroptosis; TCM intervention
缺血性脑卒中(ischemic stroke, IS)是由各种原因导致的脑组织血液供应障碍,并由此产生缺血缺氧性坏死,进而出现神经功能障礙的一组临床综合征[1]。研究显示,从1990年至2019年,卒中在我国死因中已经由第3位跃居到第1位,每年190余万人因卒中死亡[2]。在所有卒中病中,70%以上为IS[2]。目前,已知IS后导致神经元损伤和神经功能缺陷的病理机制主要包括钙超载、兴奋性氨基酸毒性、自由基损伤、炎症因子损伤、凋亡等[3]。目前,被运用于治疗脑缺血的药物、方法极为有限,主要为组织型纤溶酶原激活剂(tissue plasminogen activator, t-PA)溶栓,t-PA是唯一获得国家批准用于治疗脑缺血的药物,但需在有效时间窗内进行,否则可能会导致出血性转化,造成更严重的损伤,即脑缺血再灌注损伤[1,4]。因此,阐明脑缺血损伤机制,探索新的有效药物及作用靶点,对其治疗及预后具有重要意义。
线粒体是“细胞能量动力室”,它可以产生三磷酸腺苷(adenosine triphosphate, ATP),为机体提供能量,它在调控活性氧(reactive oxygen species, ROS)产生、调节渗透压、转导细胞信号等方面都起到关键作用[5]。当线粒体的数量、形态以及功能维持失稳态时,表现选择性受损线粒体清除及调节体系——线粒体质量控制(mitochondrial quality control, MQC)启动。MQC包括线粒体自噬、线粒体分裂/融合和线粒体生物合成,是维持线粒体稳态的重要保证[6]。
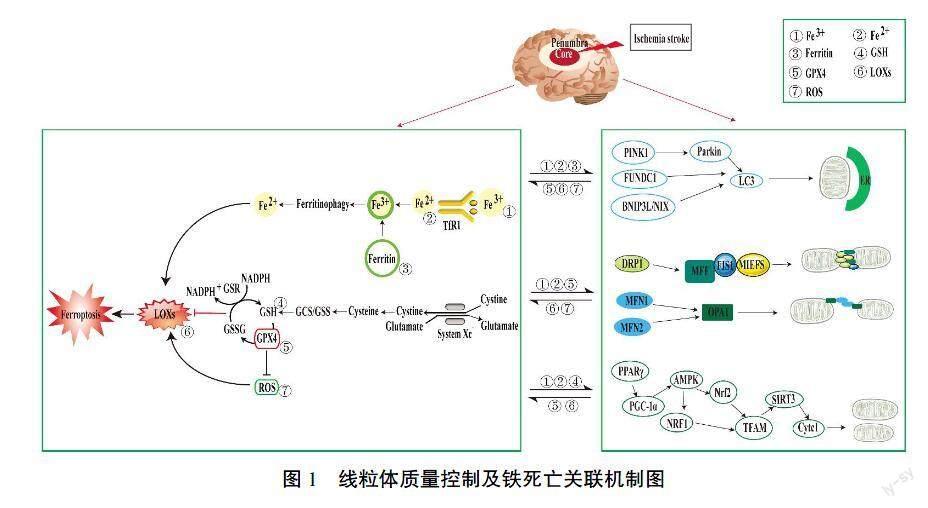
铁死亡由DIXON等[7]在cell杂志上首次提出,定义为铁依赖的、脂质过氧化物生成所致的一种程序性细胞死亡方式。当脑缺血发生后,过量的铁离子诱导脂质过氧化,线粒体膜电位降低,谷氨酸显著增多,产生神经毒性,造成神经元铁死亡,加重组织损伤[8]。线粒体作为铁利用的主要场所和氧化代谢的主要调节器,是ROS产生的主要来源,与铁死亡关系密切[5]。本文系统介绍MQC与铁死亡的关联机制,以及针对IS的研究进展进行文献综述。
1 铁死亡与MQC的关联机制研究
1.1 线粒体自噬与铁死亡
线粒体自噬是一种选择性清除多余或受损线粒体的自噬过程[9]。自噬过程中,受损细胞成分被自噬小体的双膜囊泡包裹,继而自噬小体与溶酶体融合,导致功能失调的细胞器和蛋白质降解和再循环,即自噬体-溶酶体机制[10]。在哺乳动物细胞中,线粒体自噬调节机制的经典信号通路包括PTEN诱导激酶蛋白1(PTEN-induced putative kinase1, PINK1)/E3-泛素连接酶(Parkin)依赖通路和PINK1/Parkin非依赖通路[10-11]。在脑缺血的相关研究中表明,PINK1通过自身磷酸化识别泛素,活化 Parkin包围受损的线粒体,增加其在细胞内的泛素化水平,并使其与自噬标志物微管相关蛋白1轻链3(microtubule associated protein 1 light chain3, LC3)结合,从而启动自噬过程[6,11]。非PINK1/Parkin 线粒体自噬相关信号通路包括FUN14结构域包含蛋白1(FUN14 domain-containing1, FUNDC1)、B细胞淋巴瘤2/腺病毒E1B19kDa相互作用蛋白3(B-cell lymphoma2/adenovirus E1B19-kDa-interacting protein3, BNIP3)和Nip3样蛋白X(Nip3-like protein X, NIX),以上通路蛋白可通过单独或相互作用调控线粒体自噬的发生[6]。
细胞内铁离子浓度影响线粒体自噬功能。研究表明,铁螯合剂可不依赖PINK1/Parkin通路诱导线粒体自噬[12]。铁螯合剂去铁酮(deferiprone, DFP)通过螯合铁离子增加人子宫颈癌细胞(如HeLa细胞)内铁消耗,提高铁蛋白受体(transferrin receptors, TFRC)水平,促进非PINK1/Parkin依赖的线粒体自噬[13]。非PINK1-Parkin依赖的线粒体自噬与铁代谢机制相关研究发现,PINK1突变果蝇模型中,定位于线粒体含不稳定铁硫[4Fe-4S]簇的乌头酸酶(aconitase, Acon)可增加线粒体铁蛋白表达,促进线粒体自噬[14]。另有研究表明,Parkin突变果蝇通过金属反应转录因子1(metal-responsive transcription factor1, MTF1)的过表达调节铁蛋白,改善线粒体形态,而铁螯合剂东莨菪碱磺化钠盐(bathophenanthroline sulfonated sodium salt, BPS)的干预,可促进线粒体自噬,延长Parkin突变果蝇的寿命[15-16]。
近期线粒体自噬与铁死亡的相关机制探讨,提供了疾病治疗的新靶点。有研究表明,2型糖尿病骨质疏松患者的治疗过程中,氧化磷酸化解偶联剂碳酰氰基-对-氯苯腙(carbonyl cyanide-m-chlorophenyl hydrazine, CCCP)干预可激活PINK1/Parkin通路,增强成骨细胞线粒体自噬,同时谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)减少,脂质过氧化物增多,增加细胞氧化应激,加重成骨细胞铁死亡[17]。人卵巢颗粒样肿瘤KGN细胞系经柠檬酸铁铵(ferric acid citrate, FAC)处理后,可激活转TFRC,增加铁含量,而TFRC可激活PINK1信号,诱导线粒体自噬;同时FAC上调铁摄取诱导的长链脂酰辅酶A合成酶4(acyl-CoA synthetase long chain family member 4, ACSL4),促进线粒体自噬活化和GPX4降解,并诱导脂质过氧化,进一步促进KGN细胞的铁死亡[18]。线粒体铁代谢中,NEET蛋白属于Fe-S蛋白家族,含有独特的CDGSH氨基酸序列,可参与铁硫蛋白稳态维持及氧化还原调控[19]。NEET蛋白可分为3种:CDGSH铁硫结构域1(CDGSH iron sulfur domain1, CISD1)位于线粒体外膜,CDGSH铁硫结构域2(CDGSH iron sulfur domain 2, CISD2)位于线粒体外膜及内质网,CDGSH铁硫结构域2(CDGSH iron sulfur domain3, CISD3)则局限于线粒体内[19]。研究表明,线粒体铁硫蛋白NEET基因缺失,游离铁增加,通过抑制线粒体分裂/融合,触发线粒体自噬和铁死亡,而铁螯合剂可抑制以上机制[20-21]。铁死亡诱导剂Erastin则可增加CISD3基因敲除的小鼠海马神经元细胞系(HT-22细胞)Parkin的表达,激活线粒体自噬,并促进铁死亡和脂质过氧化物的产生。因此,刺激PINK1-Parkin依赖的线粒体自噬,在CISD3蛋白介导的铁死亡中起保护作用[22]。
综上所述,研究者们提出PINK1/Parkin依赖及非依赖线粒体自噬调节通路,可能与铁死亡机制密切相关,而靶向作用于线粒体自噬调节关键蛋白能有效抑制铁死亡。
1.2 线粒体分裂/融合与铁死亡
线粒体分裂和融合是线粒体数量和形态動态变化的过程,是维持线粒体平衡的重要机制。线粒体分裂受动力相关蛋白1(dynamin-related protein1, Drp1)等的调节,而与线粒体融合相关的蛋白包括线粒体融合蛋白1(mitofusins 1, MFN1)和线粒体融合蛋白2(mitofusins 2, MFN2)[6]。线粒体结构调整需依靠GTP水解酶,其中线粒体分裂的胞质蛋白Drp1从细胞质转移至线粒体膜,诱导其普通分裂及由定位受体驱动的裂变[23];而定位于线粒体膜的MFN1/2及视神经萎缩蛋白1(optic atrophy protein1, OPA1),负责参与线粒体融合[24]。神经元氧糖剥夺/复氧(oxygen and glucose deprivation/reperfusion, OGD/R)培养中,OPA1蛋白水解增加,促进了线粒体分裂[25]。研究表明,Drp1可与3种线粒体结合蛋白相互作用,包括线粒体分裂因子(mitochondrial fission proteins, MFF)、线粒体分裂蛋白1(mitochondrial fission protein 1, FIS1)、线粒体延长因子1(mitochon?鄄drial elongation factor 1, MIEF1)和线粒体延长因子2(mitochondrial elongation factor 2, MIEF2),通过结合、缠绕、收缩,继而切断线粒体膜,导致线粒体分裂[26]。MIEF可以竞争性地减少FIS1与MFN1和MFN2的相互作用,抑制线粒体断裂,促进线粒体融合[23,26]。由此,在线粒体分裂融合机制研究中发现,MIEF可能是调节线粒体分裂和融合平衡的枢纽。
近期研究发现,线粒体的分裂/融合与铁代谢密切关联。铁离子聚集导致线粒体ROS表达增加,从而干扰线粒体分裂/融合[27]。β-地中海贫血小鼠模型中铁超载可诱导Drp1/MFN2降低,改变线粒体分裂和融合间的平衡[28]。铁离子处理后C57/BL6小鼠海马组织的Drp1表达上调[29]。研究发现,铁过载导致的游离铁水平升高能够增加人间充质基质细胞(mesenchymal stem cell, MSC)的MFF在155位丝氨酸的磷酸化(p-Ser155-MFF)水平和Drp1总蛋白水平,同时伴随着线粒体Drp1在616位丝氨酸磷酸化(p-Ser616-Drp1)水平的增加和Drp1在637位丝氨酸磷酸化(p-Ser637-Drp1)水平的降低,使Drp1由细胞质转移到线粒体外膜上,从而促进线粒体裂变[30]。铁超载增加了HT-22细胞Ca2+的表达,诱导Drp1在637位丝氨酸(Ser637-Drp1)去磷酸化,导致线粒体分裂[31]。HT-22细胞的OGD/R细胞模型中,MFN2蛋白的表达显著降低,新型铁螯合剂BHAPI处理后MFN2蛋白的表达明显增多,并起到维持铁稳态的作用[32]。脂肪细胞因子(Apelin)-13可促进线粒体钙离子单向转运体(mitochondrial calcium uniporter, MCU)的表达,导致线粒体铁超载,触发Fenton反应,诱导线粒体ROS的产生,从而增加Drp1、PINK1、Parkin的表达,促进人主动脉平滑肌细胞(human aortic vascular smooth muscle cell, HA-VSMC)的增殖[33]。抗癫痫研究中,吡仑帕奈(Perampanel)干预H2O2处理的原代神经元后,Drp1、p-Ser616-Drp1表达增加,PINK1和Parkin蛋白显著增加,铁死亡抑制相关蛋白GPX4、溶质载体家族7成员11(solute carrier family 7 member 11, SLC7A11)、铁蛋白重链多肽1(ferritin heavy chain1, FTH1)蛋白表达增加,细胞内Fe2+的水平显著降低。因此,Perampanel处理可以在氧化应激条件下调节线粒体动态变化同时,抑制铁死亡的发生[34]。
1.3 线粒体生物合成与铁死亡
线粒体生物合成是线粒体基因与细胞核基因协调表达,完成线粒体增殖及蛋白合成的一系列生物过程,包括ETC电子传递、氧化磷酸化及ATP生成等[35]。线粒体生物合成由线粒体DNA(mitochondrial DNA, mtDNA)的复制、转录、翻译与核编码蛋白的转录、翻译等多个步骤组成。脑缺血研究中,涉及线粒体生物合成相关信号通路包括过氧化物酶体增殖物激活受体γ共激活剂1-α(peroxisome proliferator-activated receptor-gamma coactivator-1alpha,PGC-1α)、AMP活化蛋白激酶(AMP-activated protein kinase, AMPK)、核呼吸因子1/2(nuclear respiratory factor1/2, NRF1/2)、线粒体转录因子A(mitochondrial transcription factor A, TFAM)及沉默信息调节因子1(sirtuin-1, SIRT1)[6,24]。神经元糖剥夺培养研究中,过氧化物酶体增殖物激活受体(peroxisome proliferator-activated receptor-gamma, PPARγ)可上调PGC-1α、NRF1、TFAM、MFNs和细胞色素C(Cytochrome C, Cyt-C)氧化酶亚基Ⅰ和Ⅳ的表达[36]。NRF1/2作为PGC-1α下游转录因子靶标,参与干预mtDNA编码的线粒体蛋白的表达。TFAM基因则包含NRF1/2的共识结合序列,能与mtDNA的D环区域启动子序列特异性结合,是mtDNA转录和复制所必需因子[37]。
铁代谢与线粒体生物合成相关性研究中发现,铁离子浓度可干预线粒体生物合成。如铁超载促进β-地中海贫血小鼠的PGC-1α表达,增加其线粒体生物合成[28]。铁螯合剂去铁胺(deferoxamine, DFO)处理后,C2C12小鼠成肌细胞因缺铁可导致PGC-1α介导的线粒体生物合成减少[38]。铁缺乏通过减少核编码线粒体基因乙酰化,对线粒体生物合成进行表观遗传调节[39]。癌症研究中发现,干预线粒体生物合成相关蛋白通路可调节铁死亡,转录调节因子核蛋白1(nuclear protein-1, NUPR1)基因抑制剂ZZW-115干预胰腺导管腺癌和肝细胞癌来源的细胞株(PDAC细胞和HCC细胞),可降低谷胱甘肽(glutathione, GSH)和GPX4活性,同时下调线粒体生物合成的关键蛋白TFAM的表达,导致脂质过氧化物聚集及细胞铁死亡[40]。胰腺癌治疗研究中发现,免疫缺陷病毒感染的抗病毒药物Zalcitabine可通过促进TFAM降解,干预人胰腺癌细胞PANC-1和Capan2细胞脂质过氧化物积聚与mtDNA应激,触发自噬依赖性铁死亡[41]。由此,铁代谢异常(铁超载、缺铁)均能干预线粒体生物合成,而其中TFAM通路蛋白可能是线粒体生物合成与铁死亡的关键调节因子。
由上可知,MQC与铁死亡联系密切。基于MQC调节,维持线粒体形态相对稳态,减轻线粒体功能损伤,可起到抗铁死亡的作用(见表1)。
2 IS线粒体质量控制异常与神经元铁死亡的关联研究
2.1 IS中MQC與铁死亡关联机制
脑卒中即中风,是常见的急性脑血管病。到2019年,我国新发脑卒中病例高达3 940万,发病率较1990年上升了86%,死亡率上升了32.3%,是世界第二大死亡原因之一,而所有中风病中,70%以上为IS[42]。随着IS发病率和致残率的持续上升,研究者们在更积极探索其病理生理新机制。
研究者们发现,IS导致线粒体自噬、线粒体动力学异常和线粒体生物合成受损[43]。同时,脑缺血诱发神经元铁死亡,而铁螯合剂(例如去铁胺、环吡罗司和去铁酮)可通过减少细胞内游离铁离子抑制铁死亡[44-45]。有研究提出,抑制线粒体损伤作为调节铁依赖性神经元铁死亡的机制,可能是IS的潜在治疗策略[46]。铁死亡诱导剂RSL3导致神经元HT22细胞线粒体碎片化、线粒体膜电位丧失和减少线粒体呼吸,而ROS清除剂MitoQ可起到抑制线粒体形态及功能损伤,保护神经元作用[47]。线粒体融合蛋白(Mitofusin, MFN)是发动蛋白(Dynamin)超家族中重要一员,对线粒体融合/分裂的结构形态具有重要的调节作用[48]。同时,有研究者提出Dynamin抑制剂Dynasore可通过抑制线粒体呼吸及脂质过氧化物的产生,减轻脑缺血后铁死亡导致的神经元损伤[48]。脑缺血后,促进线粒体生物合成相关的转录激活剂NRF2可介导调节神经元铁死亡[49]。由此可见,脑卒中后MQC与铁死亡的关联机制研究仍处于起步阶段,揭示MQC与铁死亡发生、发展的相关病理机制,可为IS防治提供重要的分子靶点。以上概述见图1。
2.2 中医药靶向线粒体质量控制及铁死亡治疗IS的研究
中医药在IS临床应用中有着独到的优势,它的作用机制与多方面有关,具体包括抗兴奋性氨基酸、抗自由基损伤、抑制钙超载、调节细胞自噬、调节凋亡、调节炎症反应、促进血管新生和神经修复等[50]。在中医理论中,“气”是组成并维系着人体生命活动的基础[51]。气化可促使精、气、血、津液等物质的新陈代谢及相互转化[52]。中医学认为气是万物生化本源[52-53]。现代医学认为,线粒体是细胞的“能量工厂”[54],线粒体进行能量合成与中医的气有一定相似之处[53,55]。中医学认为,“精”“气”“血”“津”“液”是构成机体的基础,也是维持生命活动的基本物质。其中,“精”是组成人体的最基础的物质,也是保持身体健康的重要因素。“气”是从“精”中产生的极其细小的物质,再和肺部所吸收的天地间清气结合在一起形成的[55-56]。生理学提出线粒体为“能量工厂”,是细胞内氧化磷酸化和ATP生成的主要场所[54]。有研究者提出,“气”与ATP化生同源,“气”与线粒体功能统一[52,56-57]:(1)机体吸入的氧气和消化吸收的小分子物质参与线粒体的氧化磷酸化反应,产生ATP;中医理论中,氧气和通过肺吸收的自然清气有很大的关系,而这些小分子物质就是由脾胃运化成的水谷精微,也就是谷气。所以,“气”与ATP都来自于同一种物质,其来源也是同一种物质。(2)中医学理论中“气”可推动与调控、温煦与凉润、防御与固摄;而线粒体提供ATP,是生命活动能量的直接来源,参与全身器官系统防御和调节等各生理功能。由此,中医学中的气与线粒体的内涵相关及功能统一。王清任在《医林改错》中认为“元气既虚,必不能达于血管,血管无气,必停留而瘀”,表明是由于气血不足而造成血瘀[58-59]。因此,中医学认为“气虚血瘀”是脑卒中的重要发病机制。血液的流动离不开气的作用,在中风的发生过程中,气虚是其发病基础,而血瘀是其疾病产物[60],故治疗多用益气活血中药及其单体[59]。近年来,研究者们开始探讨中药通过调节MQC及抑制铁死亡减轻脑缺血损伤的新机制。
有关线粒体自噬的研究表明,黄芩苷能有效調控缺氧再灌注PC12细胞模型线粒体自噬相关蛋白BNIP3、Parkin、FUNDC1的mRNA表达,可通过干预线粒体自噬减轻神经元损伤[61];而黄芩苷抑制铁死亡的治疗机制,是通过干预二价金属转运蛋白(divalent metal transporter1, DMT1),上调溶质转运蛋白家族3成员2(Solute carrier family 3 member 2, SLC3A2)、GPX4的表达实现[62]。在干预脑缺血再灌注小鼠线粒体质量控制机制研究中,白藜芦醇可激活PINK1和Parkin蛋白的表达,促进线粒体自噬[63-64],也能通过活化AMPK/PGC-1α/TFAM通路,增加mtDNA拷贝数,促进线粒体生物合成[63]。另有研究报道,白藜芦醇干预可上调脑出血模型大鼠GPX4蛋白表达,减少脂质氧化产物丙二醛(malondialdehyde, MDA)生成,抑制神经元铁死亡[65]。
线粒体动力学的研究表明,活血祛瘀代表方补阳还五汤可下调缺血再灌注脑损伤大鼠Drp1、FIS1、Cyt-C的表达水平,抑制线粒体分裂,减轻大鼠海马神经元缺氧复氧损伤[66];网络药理学筛选PC12细胞氧化应激模型的相关铁死亡靶点并进行验证,发现补阳还五汤可能通过热休克蛋白90(heat shock protein 90, HSP90)、表皮生长因子受体(epidermal growth factor receptor, EGFR)、生长因子受体结合2(growth factor receptor-bound protein 2,GRB2)等靶点及控丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)、磷脂酰肌醇3激酶/蛋白激酶B(phosphatidylinositol 3 kinase/protein kinase B, PI3K/AKT)等信号通路抑制铁死亡,缓解脑缺血的继发性损伤[67]。丹参酮ⅡA干预大鼠局灶性脑缺血再灌注后,可改善缺血区脑血流量、下调Drp1及半胱氨酸天冬氨酸蛋白酶3(cysteinyl aspartate specfic protease 3, Caspase-3)表达,起到抑制线粒体分裂及神经元凋亡的作用[68];铁代谢调节研究发现,丹参酮ⅡA可抑制脑缺血后铁死亡,其作用机制包括调节铁稳态,抑制脂质过氧化物的生成[69],以及下调DMT1,增加膜铁转运蛋白(ferroportin1, FPN1)表达,减少铁聚集[70]。
線粒体生物合成的相关研究发现,三七总皂苷对6-羟基多巴胺诱导的人神经母细胞瘤SH-SY5Y细胞株损伤具有一定的保护作用,该保护机制可能与激活Nrf2、抑制氧化应激和促进线粒体生成有关[71];同时,三七总皂苷可显著降低局灶性脑缺血大鼠炎症因子白细胞介素-1β(interleukin-1β, IL-1β)、肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)和白细胞介素-6(interleukin-6, IL-6)的表达,抑制游离二价铁离子和脂质过氧化终产物MDA的表达,增加GSH及GPX4水平,通过干预炎症和抑制铁死亡保护神经元[72]。
脑卒中的中药治疗具有多靶点、多通路的干预特征。研究发现,益气活血中药复方及单体可有效调节IS后线粒体质量控制,抑制神经元铁死亡。进一步研究探讨两者在脑卒中后的关联机制,可为IS的治疗提供新思路,为药物干预提供关键靶点。以上概述见表2。
3 前景与展望
线粒体是糖类、脂肪和氨基酸通过三羧酸循环与氧化磷酸化过程释放能量的场所,被称为“能量工厂”。MQC包括线粒体自噬、线粒体分裂/融合及线粒体生物合成。铁离子是铁硫蛋白的合成底物,既可作为电子传递蛋白质的辅基,参与能量转移,又可作为某些酶的活性基团,参与各种生化反应。MQC与铁死亡关联机制的研究可为疾病防治提供新的治疗靶点。IS后MQC异常,同时缺血诱发神经元铁死亡。关于IS后MQC与铁死亡损伤机制关联靶点的研究鲜有报道;中药防治IS具有独特优势,其作用机制涉及多方面,而干预IS后MQC及铁死亡的研究多处于起步阶段。因此,探讨IS发生后MQC及铁死亡关联病理机制,可为药物防治研究拓宽新方向,也为筛选新的有效中药提供重要的分子靶点和技术平台。
参考文献
[1] RABINSTEIN A A. Update on treatment of acute ischemic stroke[J]. Continuum, 2020, 26(2): 268-286.
[2] 王陇德, 彭 斌, 张鸿祺, 等. 《中国脑卒中防治报告2020》概要[J]. 中国脑血管病杂志, 2022, 19(2): 136-144.
[3] KHOSHNAM S E, WINLOW W, FARZANEH M, et al. Pathogenic mechanisms following ischemic stroke[J]. Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, 2017, 38(7): 1167-1186.
[4] FUKUTA T, ASAI T, YANAGIDA Y, et al. Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke[J]. The FASEB Journal, 2017, 31(5): 1879-1890.
[5] WANG H, LIU C, ZHAO Y X, et al. Mitochondria regulation in ferroptosis[J]. European Journal of Cell Biology, 2020, 99(1): 151058.
[6] TIAN H Y, CHEN X Y, LIAO J, et al. Mitochondrial quality control in stroke: From the mechanisms to therapeutic potentials[J]. Journal of Cellular and Molecular Medicine, 2022, 26(4): 1000-1012.
[7] DIXON S J, LEMBERG K M, LAMPRECHT M R, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death[J]. Cell, 2012, 149(5): 1060-1072.
[8] G?魣MEZ A, ALVA N, CARBONELL T, et al. Extracellular ferritin contributes to neuronal injury in an in vitro model of ischemic stroke[J]. Journal of Physiology and Biochemistry, 2021, 77(4): 539-545.
[9] QIU Y H, ZHANG T S, WANG X W, et al. Mitochondria autophagy: A potential target for cancer therapy[J]. Journal of Drug Targeting, 2021, 29(6): 576-591.
[10] YANG J L, MUKDA S, CHEN S D. Diverse roles of mitochondria in ischemic stroke[J]. Redox Biology, 2018, 16: 263-275.
[11] WU H, WANG F L, TA N, et al. The multifaceted regulation of mitochondria in ferroptosis[J]. Life, 2021, 11(3): 222.
[12] ALLEN G F G, TOTH R, JAMES J, et al. Loss of iron triggers PINK1/parkin-independent mitophagy[J]. EMBO Reports, 2013, 14(12): 1127-1135.
[13] WILKINSON K A, GUO C. Iron chelation promotes mitophagy through SENP3-mediated deSUMOylation of FIS1[J]. Autophagy, 2022, 18(7): 1743-1745.
[14] ESPOSITO G, VOS M, VILAIN S, et al. Aconitase causes iron toxicity in Drosophila pink1 mutants[J]. PLoS Genetics, 2013, 9(4): e1003478.
[15] SAINI N, GEORGIEV O, SCHAFFNER W. The parkin mutant phenotype in the fly is largely rescued by metal-responsive transcription factor (MTF-1)[J]. Molecular and Cellular Biology, 2011, 31(10): 2151-2161.
[16] SAINI N, OELHAFEN S, HUA H Q, et al. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways[J]. Neurobiology of Disease, 2010, 40(1): 82-92.
[17] WANG X D, MA H D, SUN J, et al. Mitochondrial ferritin deficiency promotes osteoblastic ferroptosis via mitophagy in type 2 diabetic osteoporosis[J]. Biological Trace Element Research, 2022, 200(1): 298-307.
[18] ZHANG L Z, WANG F, LI D M, et al. Transferrin receptor-mediated reactive oxygen species promotes ferroptosis of KGN cells via regulating NADPH oxidase 1/PTEN induced kinase 1/acyl-CoA synthetase long chain family member 4 signaling[J]. Bioengineered, 2021, 12(1): 4983-4994.
[19] MITTLER R, DARASH-YAHANA M, SOHN Y S, et al. NEET proteins: A new link between iron metabolism, reactive oxygen species, and cancer[J]. Antioxidants & Redox Signaling, 2019, 30(8): 1083-1095.
[20] NECHUSHTAI R, KARMI O, ZUO K, et al. The balancing act of NEET proteins: Iron, ROS, calcium and metabolism[J]. Biochimica et Biophysica Acta Molecular Cell Research, 2020, 1867(11): 118805.
[21] PARK J, LEE D G, KIM B, et al. Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells[J]. Toxicology, 2015, 337: 39-46.
[22] LI Y C, WANG X, HUANG Z H, et al. CISD3 inhibition drives cystine-deprivation induced ferroptosis[J]. Cell Death & Disease, 2021, 12: 839.
[23] VONGSFAK J, PRATCHAYASAKUL W, APAIJAI N, et al. The alterations in mitochondrial dynamics following cerebral ischemia/reperfusion injury[J]. Antioxidants, 2021, 10(9): 1384.
[24] 魏 旋, 劉吉勇, 张文丽, 等. 靶向线粒体质量控制防治脑缺血再灌注损伤的研究进展[J]. 中国中药杂志, 2022, 47(16): 4305-4313.
[25] SANDERSON T H, RAGHUNAYAKULA S, KUMAR R. Neuronal hypoxia disrupts mitochondrial fusion[J]. Neuroscience, 2015, 301: 71-78.
[26] YU R, LIU T, JIN S B, et al. MIEF1/2 orchestrate mitochondrial dynamics through direct engagement with both the fission and fusion machineries[J]. BMC Biology, 2021, 19(1): 229.
[27] SABOUNY R, SHUTT T E. Reciprocal regulation of mitochondrial fission and fusion[J]. Trends in Biochemical Sciences, 2020, 45(7): 564-577.
[28] KHAMSEEKAEW J, KUMFU S, WONGJAIKAM S, et al. Effects of iron overload, an iron chelator and a T-Type calcium channel blocker on cardiac mitochondrial biogenesis and mitochondrial dynamics in thalassemic mice[J]. European Journal of Pharmacology, 2017, 799: 118-127.
[29] 王 鲜. 铁暴露与认知功能损伤的关联及作用机制研究[D]. 武汉: 华中科技大学, 2019.
[30] ZHENG Q Q, ZHAO Y S, GUO J, et al. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway[J]. Cell Death & Disease, 2018, 9: 515.
[31] LEE D G, PARK J, LEE H S, et al. Iron overload-induced calcium signals modulate mitochondrial fragmentation in HT-22 hippocampal neuron cells[J]. Toxicology, 2016, 365: 17-24.
[32] CHEN X L, ZHANG G P, GUO S L, et al. Mfn2-mediated preservation of mitochondrial function contributes to the protective effects of BHAPI in response to ischemia[J]. Journal of Molecular Neuroscience, 2017, 63(3): 267-274.
[33] 楊 霄. 线粒体自噬引起线粒体铁超载诱导Warburg效应介导Apelin-13促人主动脉血管平滑肌细胞增殖[D]. 衡阳: 南华大学, 2020.
[34] 艾 璞. Perampanel通过线粒体自噬缓解过氧化氢诱导神经元氧化应激下铁死亡的作用机制[D]. 合肥: 安徽医科大学, 2022.
[35] POPOV L D. Mitochondrial biogenesis: An update[J]. Journal of Cellular and Molecular Medicine, 2020, 24(9): 4892-4899.
[36] MIGLIO G, ROSA A C, RATTAZZI L, et al. PPARγ stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss[J]. Neurochemistry International, 2009, 55(7): 496-504.
[37] CARDANHO-RAMOS C, MORAIS V A. Mitochondrial biogenesis in neurons: How and where[J]. International Journal of Molecular Sciences, 2021, 22(23): 13059.
[38] RENSVOLD J W, ONG S E, JEEVANANTHAN A, et al. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis[J]. Cell Reports, 2013, 3(1): 237-245.
[39] RENSVOLD J W, KRAUTKRAMER K A, DOWELL J A, et al. Iron deprivation induces transcriptional regulation of mitochondrial biogenesis[J]. Journal of Biological Chemistry, 2016, 291(40): 20827-20837.
[40] HUANG C, SANTOFIMIA-CASTA?譙O P, LIU X, et al. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner[J]. Cell Death Discovery, 2021, 7: 269.
[41] LI C F, ZHANG Y, LIU J, et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death[J]. Autophagy, 2021, 17(4): 948-960.
[42] MA Q F, LI R, WANG L J, et al. Temporal trend and attributable risk factors of stroke burden in China, 1990-2019: An analysis for the Global Burden of Disease Study 2019[J]. The Lancet Public Health, 2021, 6(12): e897-e906.
[43] MORRIS G, BERK M, CARVALHO A F, et al. Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases[J]. Behavioural Brain Research, 2018, 341: 154-175.
[44] STOCKWELL B R, FRIEDMANN ANGELI J P, BAYIR H, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease[J]. Cell, 2017, 171(2): 273-285.
[45] AGRAWAL S, FOX J, THYAGARAJAN B, et al. Brain mitochondrial iron accumulates in Huntingtons disease, mediates mitochondrial dysfunction, and can be removed pharmacologically[J]. Free Radical Biology and Medicine, 2018, 120: 317-329.
[46] HUANG Y, LIU J Y, HE J L, et al. UBIAD1 alleviates ferroptotic neuronal death by enhancing antioxidative capacity by cooperatively restoring impaired mitochondria and Golgi apparatus upon cerebral ischemic/reperfusion insult[J]. Cell & Bioscience, 2022, 12(1): 42.
[47] JELINEK A, HEYDER L, DAUDE M, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis[J]. Free Radical Biology and Medicine, 2018, 117: 45-57.
[48] CLEMENTE L P, RABENAU M, TANG S, et al. Dynasore blocks ferroptosis through combined modulation of iron uptake and inhibition of mitochondrial respiration[J]. Cells, 2020, 9(10): 2259.
[49] RATAN R R. The chemical biology of ferroptosis in the central nervous system[J]. Cell Chemical Biology, 2020, 27(5): 479-498.
[50] 李兆珍, 張丹参. 脑缺血再灌注损伤相关机制的研究进展[J]. 神经药理学报, 2020, 10(6): 60-63.
[51] 郑鸿桥. 概念辨类与译法择取: 中医术语“气”的英译再思考[J]. 广西中医药大学学报, 2019, 22(2): 143-147.
[52] 林 飞, 郭丽丽, 王 阶. 基于线粒体的功能阐释中医“气”的作用[J]. 中国中西医结合杂志, 2014, 34(8): 903-906.
[53] 张永忠. 论中医学人体之气的实质是新陈代谢[J]. 中国中医基础医学杂志, 2000, 6(5): 8-11.
[54] TRIGO D, AVELAR C, FERNANDES M, et al. Mitochondria, energy, and metabolism in neuronal health and disease[J]. FEBS Letters, 2022, 596(9): 1095-1110.
[55] 张茂林, 张六通, 邱幸凡, 等. 论线粒体与中医“气”的关系[J]. 中国中医基础医学杂志, 2001, 7(4): 60-61.
[56] 李 斌, 纪立金, 闵 寅, 等. 从《黄帝内经》的思维方法探讨“气”和能量的相关性[J]. 中华中医药杂志, 2019, 34(11): 5033-5036.
[57] 陈文为. 从生物能学探讨中医”气”的實质[J]. 北京中医药大学学报, 1994, 17(2): 7-9.
[58] 戴皓宁. 《医林改错》补气活血法初探[J]. 中国民间疗法, 2018, 26(9): 2-3.
[59] 李南方, 陈永斌, 刘启华, 等. 益气活血法治疗缺血性中风的研究进展[J]. 世界中西医结合杂志, 2020, 15(5): 974-976, 980.
[60] 刘 轲. 缺血性脑卒中中医病机探讨[J]. 江苏中医药, 2003, 35(10): 49.
[61] 林筱洁, 周惠芬, 虞 立, 等. 基于线粒体自噬调控的黄芩苷减轻神经元细胞缺血缺氧/再灌注损伤的研究[J]. 中华中医药杂志, 2019, 34(5): 2022-2027.
[62] 季小添. 黄芩苷抑制神经元铁死亡改善脑出血的作用及机制[D]. 广州: 广州中医药大学, 2020.
[63] 裴美娟, 王 恺, 张 婷, 等. 白藜芦醇对脑梗死小鼠线粒体生物合成及脑水肿的影响[J]. 遵义医学院学报, 2018, 41(2): 156-159, 164.
[64] 向 菲. 白藜芦醇对脑缺血再灌注损伤中线粒体的保护作用及相关机制研究[D]. 重庆: 重庆医科大学, 2019.
[65] 佘仁夏, 张 潘, 何晓英. 白藜芦醇对脑出血大鼠铁死亡的影响[J]. 四川医学, 2021, 42(3): 254-259.
[66] 韦 辰, 王士雷, 孔宪刚, 等. 补阳还五汤对脑缺血再灌注损伤大鼠线粒体分裂蛋白Drp1、Fis1及细胞色素C表达的影响[J]. 陕西中医, 2017, 38(10): 1481-1483.
[67] 赵冯岩, 杨浩澜, 朱炎贞, 等. 基于网络药理学从铁死亡途径研究补阳还五汤调控缺血性脑卒中的作用机制[J]. 湖南中医药大学学报, 2021, 41(7): 1065-1072.
[68] 韩若东. 丹参酮ⅡA对大鼠脑缺血再灌注后细胞凋亡、Drp-1及Caspase 3表达的影响[D]. 合肥: 安徽医科大学, 2012.
[69] 许 璐. 丹参酮ⅡA通过调节铁稳态抑制脑缺血模型中铁死亡的机制研究[D]. 合肥: 安徽医科大学, 2019.
[70] ZHOU Z Y, ZHAO W R, ZHANG J, et al. Sodium tanshinone IIA sulfonate: A review of pharmacological activity and pharmacokinetics[J]. Biomedecine & Pharmacotherapie, 2019, 118: 109362.
[71] 汪梦霞, 赵静宇, 孙冬梅, 等. 三七总皂苷对6-羟基多巴胺诱导SH-SY5Y细胞损伤的保护作用及可能机制[J]. 药学学报, 2016, 51(6): 898-906.
[72] 王林琳, 康智能, 刘文鹏, 等. 三七总皂苷抑制铁死亡和炎症反应减轻大鼠脑缺血再灌注损伤[J]. 中国免疫学杂志, 2022, 38(3): 296-300.
