Nrf2调控的铁死亡途径在非酒精性脂肪性肝病防治中的作用机制*
2023-08-14貟志强姚婷婷衣雪洁
貟志强 姚婷婷 李 涛 衣雪洁
(1)沈阳体育学院运动人体科学学院,沈阳 110115;2)沈阳体育学院实验室管理中心,沈阳 110115;3)沈阳体育学院运动与健康研究中心,沈阳 110115)
随着肥胖的流行,非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)患病率逐年增加,目前被认为是全球最普遍的慢性肝病之一[1-2]。NAFLD是一种进展性肝病,最初的特点是单纯的脂肪变性,进而发展成非酒精性脂肪性肝炎(nonalcoholic steatohepatitis,NASH)、肝纤维化甚至肝细胞癌[3]。全球约25% 的成年人患有NAFLD,中国NAFLD 的总体患病率约为30%[4]。由于高发病率和潜在的严重危害,NAFLD 已成为重要的公共健康问题之一。但是NAFLD的发病机制较为复杂,目前尚未完全阐明。“二次打击学说”是NAFLD 发病的经典机制,目前将胰岛素抵抗引起的肝脏脂肪变性认为是“第一次打击”;而活性氧(reactive oxygen species,ROS)堆积导致的炎症、内质网应激和氧化应激被认为是“第二次打击”,氧化应激在其中起着关键作用[5-7]。巧合的是,铁死亡的重要特征之一也是脂质过氧化物堆积以及抗氧化酶谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)减少。为此一些学者开始探究铁死亡与NAFLD的关系及其作用的机制,发现引起铁死亡的多种机制,也参与了NAFLD 的发生发展[3,8-9]。其中,核因子E2 相关因子2(nuclear factor E2 related factor 2,Nrf2) 在铁死亡参与NAFLD 的发展中扮演重要的角色[9-11],本文对Nrf2、铁死亡和NAFLD 的关系进行梳理,总结Nrf2 通过铁死亡调控NAFLD 的可能机制,并提出目前研究可能存在的问题,为防治NAFLD 提供了新的视野。
1 非酒精性脂肪性肝病(NAFLD) 与铁死亡
2012 年Dixon 等[12]首次提出铁死亡是一种新型程序性死亡,它是指由于铁依赖性的脂质过氧化物堆积而导致的非凋亡形式的细胞死亡。其形态学特征区别于凋亡、坏死和自噬,主要表现为线粒体体积减小、膜密度增加、线粒体嵴减少或消失。已有研究表明,过度的铁死亡与神经退行性疾病[13]、缺血/再灌注诱导的器官损伤[14]、心肌梗塞[15]和NAFLD[16]有因果关系,抑制铁死亡可能对这些疾病起到改善效果。另外,抑制铁死亡有助于抑制多种癌症的发展[17]。
肝脏是铁储存、脂质代谢的重要器官,肝细胞中铁代谢异常和脂质过氧化物的过度积累都会引起铁死亡[18]。2019 年日本Minoru Tanaka 研究小组[19]首次发现,NASH 模型小鼠肝细胞中铁死亡先于其他的细胞死亡。特异性抑制脂质过氧化反应可抑制铁死亡的发生,减缓肝脏损伤。之后的学者进一步发现,GPX4、铁代谢、脂质过氧化以及一些其他途径参与了肝脏铁死亡的调控,改善肝脏过度铁死亡可能成为防治NAFLD的重要方法之一。
1.1 NAFLD与铁死亡的GPX4途径
脂质过氧化物堆积以及GPX4减少是铁死亡的主要特征之一[20-21]。在正常状态下,GPX4可通过其辅因子谷胱甘肽(glutathione,GSH),将有毒的脂质氢过氧化物(L-OOH)转化为无毒的脂质醇(L-OH),从而清除脂质过氧化物[22]。因此,抑制GPX4会导致脂质过氧化物积累,这也是铁死亡的标志之一[23]。但在NAFLD 的不同发展阶段GPX4的变化不尽相同。
近几年的研究显示,肝脏单纯性脂肪变性不仅有GPX4 的表达下降,同时伴随着铁死亡的发生,在这时增加GPX4 的表达不仅可以负向调节铁死亡,对NAFLD 的发展也可以起到有效的抑制作用[9]。高脂饮食(HFD)的NAFLD模型或棕榈酸/油酸(PA/OA)诱导的肝细胞变性模型中均显示GPX4表达降低,且发生铁死亡[3]。肝细胞中沉默GPX4 后脂质变性加剧,通过激活胸腺肽β4(thymosin beta 4,Tβ4)来增加GPX4 表达可抑制铁死亡,改善NAFLD[8]。
NAFLD 得不到有效的控制,可进展为危害性更大的NASH,但是NAFLD 如何发展为NASH 尚不清楚。不同于NAFLD,在NASH 阶段,GPX4表达量显著升高。对C57BL/6小鼠进行3周蛋氨酸/胆碱缺乏饮食(MCD)饮食干预后,发现小鼠肝脏GPX4 表达显著增加,并伴随着铁死亡,ROS、肝Fe(II)和总铁增加,脂质过氧化物合成酶酰基辅酶A 合成酶长链家族成员4 (acyl-CoA synthetase long-chain family member 4,ACSL4)和脂氧合酶(lipoxygenase,ALOX)显著增加,同时发生NASH[24]。另一篇使用C57BL/6 小鼠通过MCD 干预10 d 制备小鼠NASH 模型,同样出现了GPX4表达量显著升高和铁死亡加剧的现象[25]。这提示,GPX4 在NASH 中升高,这与单纯性脂肪肝中GPX4 下降刚好相反。可以推测,随着NAFLD病程的加剧,会出现严重的炎症及脂质过氧化反应,机体为了保护细胞免受危害会做出应激反应,这可能是NASH中GPX4表达上升的原因,但上升的GPX4并不能完全清除过多的脂质过氧化物,从而发生铁死亡。
然而,造模时间的不同可能对GPX4 在NASH中的功能产生影响。一项研究发现,对C57BL/6小鼠进行MCD 饮食干预4、8、12 周,NASH 严重程度随着时间的增加而增加,同时伴有GPX4的持续上升, 不同的是 GSH 升高和丙二醛(malondialdehyde,MDA)持续下降,提示铁死亡可能受到抑制。这与之前研究中铁死亡促进NASH进展相悖。作者认为,防治NASH的进展中,铁死亡可能作为一种修复或抵抗调节剂来减轻NASH中的肝损伤。离体实验显示,对人正常肝细胞(LO2)给予高游离脂肪酸(HFFA)72 h 建造的NASH 模型,在过表达GPX4 时,LO2 细胞脂质积累增加,在使用RSL3后脂质积累减轻,这与之前研究结果相反[26]。推测,早期NASH 中,增加GPX4 表达可抑制铁死亡改善NASH。但这篇文章对C57BL/6小鼠MCD饮食干预4、8、12周显著高于其他研究报道的3周和10 d,并且离体实验中通过HFFA对LO2细胞进行72 h NASH造模,也显著高于24 h PA 处理的LO2 细胞[8]。长时间的NASH造模可能导致肝炎达到不可逆的程度,造成脂质堆积持续性增加。并且文章只对离体实验中GPX4表达进行干预,无法验证在体实验中长时间NASH造模后,增加GPX4 表达是否也发挥脂质沉积的作用。这一结果需要后续研究验证不同造模时间对GPX4 与脂质积累的影响。且在NASH 模型中,过表达烯醇化酶3(ENO3)会引起GPX4 表达增加,同时脂质积累加重。有学者已证明,ENO3可加速胆固醇酯合成引起肝脏胆固醇酯积累[27]。提示ENO3 可能通过GPX4 调控了肝脏的胆固醇合成与积累,但相关研究尚浅,还需要进一步的验证。
上述研究结果虽然不尽一致,但大多结果表明,GPX4 下降与铁死亡和NAFLD 有关,升高GPX4 能抑制铁死亡,改善NAFLD。但在不同程度的NASH状态下,肝细胞GPX4表达增加与铁死亡和NASH 病程的关系结果并不一致,铁死亡在NASH发展中的作用机制还有待进一步的研究。
1.2 NAFLD与铁死亡的铁代谢途径
铁稳态失衡不仅是许多疾病(动脉粥样硬化、心肌病、肾缺血再灌注和神经退行性疾病)的诱发因素之一[28],也是NAFLD 的病因之一[29]。肝脏是机体铁储存的主要器官,对于维持机体铁稳态十分重要。膳食铁在肠细胞中被血红素加氧酶(hemeoxygenase,HO-1)降解,分解为Fe(II)、胆绿素和胆红素。Fe(II)通过细胞膜铁转运蛋白(ferroportin,FPN)转运入血液,通过血液循环维持机体铁稳态。在肝细胞中,血液中的铁通过与肝细胞膜上的转铁蛋白受体1(transferrin receptor 1,TFR1)结合,内吞进入细胞。Fe(III)在酸性环境中从转铁蛋白中释放出来,通过前列腺六跨膜上皮抗原3 (six-transmembrane epithelial antigen of the prostate 3,STEAP3)还原为不稳定的高反应性Fe (II),一部分通过二价金属转运蛋白1(divalent metal transporter 1,DMT1)和瞬时受体电位阳离子通道黏脂蛋白1/2(transient receptor potential mucolipin 1/2,TRPML1/2)转运到溶酶体膜中,进入细胞内不稳定铁池(labile iron pool,LIP),另一部分Fe(II)可被铁蛋白吸收。另外,肝细胞还可以通过分泌铁调素负反馈调节FPN 阻止铁流出细胞进入血液循环中,以维持机体整体铁稳态。
当机体铁稳态受到破坏时,过量的Fe(II)会导致铁过载的发生,这不仅是铁死亡的诱发因素之一,也是NAFLD的发病原因之一[29]。研究人员为证明过量的铁离子在NAFLD中的作用,通过HFD建立NAFLD小鼠模型,发现在小鼠肝脏中总铁浓度无显著变化,但Fe(II)浓度升高[9]。值得注意的是,由LIP 释放的Fe(II)是铁过载和芬顿反应发生的原因之一。在发生NAFLD条件下,过量的铁会产生具有氧化还原活性的非转铁蛋白结合铁(nontransferrin-binding iron,NTBI),其摄取由金属转运蛋白(metal transporter Zip14,SLC39A14)介导。通常情况下,铁储存在肝脏铁蛋白中,而铁过载时核受体共激活因子4 (nuclear receptor coactivator,NCOA4)与铁蛋白结合,将其输送至溶酶体,导致LIP中铁积累加剧,最终释放大量的Fe (II)[30]。Fe (II) 与过氧化氢(hydrogen peroxide,H2O2)反应生成Fe(III)与羟基自由基(HO·),发生芬顿反应[31-32]。芬顿反应产生的HO·攻击膜上的多不饱和脂肪酸导致脂质过氧化物的生成增加,进而引发铁死亡。这提示NAFLD 中Fe(II)的增多可能是铁死亡的重要原因[9](图1)。同样Li等[24]发现,NASH 小鼠肝脏同样出现铁过载,通过铁螯合剂DFO 缓解了这一现象,并且抑制铁死亡,改善了NASH。以上结果表明,铁过载引发的脂质过氧化物增加以及铁死亡的发生可能是导致小鼠发生NAFLD的重要原因,降低铁过载可能对NAFLD的治疗起到重要作用。
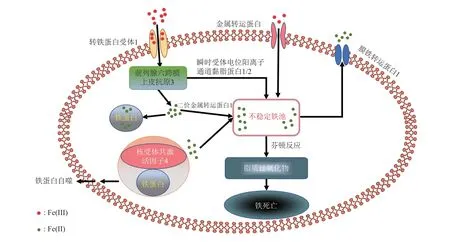
Fig.1 Iron metabolism in hepatocytes图1 肝细胞中的铁代谢
相反,有研究显示未出现铁过载情况下,也会发生铁死亡与肝损伤。多聚胞嘧啶RNA结合蛋白1(polyr(C)binding protein 1,PCBP1)是一种多功能蛋白质,可作为胞质铁伴侣,与铁结合并转移至哺乳动物细胞中的受体蛋白[33]。LIP中95%是由PCBP1 与Fe-GSH 复合物组成,而Fe-GSH 是由GSH 通过其游离巯基直接配位Fe(II)形成[34]。有研究报道,在PCBP1 敲除小鼠中肝细胞铁水平降低,而LIP中的具有高反应性的铁含量增加,其中未与PCBP1 结合的Fe(II)导致ROS 产生,在不出现铁过载的情况下导致脂质过氧化物增加,加剧脂质变性。辅酶Q10 可逆转PCBP1 敲除引起的ROS 增加以及肝脂肪变性[35]。因此,在肝脏中PCBP1 可能通过调控LIP,从而抑制铁死亡改善NAFLD。
综上所述,铁过载是NAFLD 发生的重要原因,也是引发铁死亡的关键因素。因此降低铁过载抑制铁死亡对于NAFLD 的发展可能起到缓解作用。另外,PCBP1 可能成为降低铁过载的可行靶标。
1.3 NAFLD与铁死亡的脂质过氧化途径
NAFLD 的发病机制中,脂质过氧化物积累引起的氧化应激被认为是一个重要的因素[36],而脂质过氧化物积累也是铁死亡发生的原因之一[3]。因此,脂质过氧化可能是引发铁死亡,导致NAFLD发展的关键因素。
脂质过氧化物的生成是由包括花生四烯酸(arachidonic acid,AA)在内的多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs),通过ACSL4的催化下生成多不饱和脂肪酸- 辅酶A(polyunsaturated fatty acid,PUFA-CoA),随后在溶 磷 脂 酰 胆 碱 酰 基 转 移 酶 3(lysophosphatidylcholine acyltransferase 3,LPCAT3)催化下形成多不饱和脂肪酸-磷脂酰乙醇胺 (poly-unsaturated fatty acid-phosphatidyl ethanolamine,PUFA-PE),最终通过酶促或非酶促(芬顿反应)生成脂质过氧化物[37]。近来报道,HFD 诱导的NAFLD 小鼠中发现脂质过氧化产物MDA 以及Fe(II)含量显著增加,同时引发铁死亡[9]。在NASH小鼠中,脂质过氧化底物AA的上升是最明显的过程之一。并在小鼠肝脏中发现铁过载、MDA 增加以及脂质堆积的现象,使用铁死亡抑制剂后脂质堆积与铁过载得到改善,脂质过氧化产物MDA 下降,NASH 程度下降[24]。上文讨论过,在铁过载时,过多的Fe(II)可能引发芬顿反应,参与脂质过氧化反应。因此,参与脂质过氧化物生成的AA 与Fe(II)在脂质过氧化反应中可能存在协同关系,共同增加铁死亡的发生,加速NAFLD的进展。
ACSL4 是PUFA 生成脂质过氧化物的关键酶,可将PUFA 氧化为5- 羟基二十碳四烯酸(5-hydroxyeicosatetraenoic acid, 5-HETE)。 Wei等[38]将大鼠暴露于砷,建立NASH 以及铁死亡模型,结果显示ACSL4 和MDA表达明显升高。抑制ACSL4 表达后,由砷诱导的大鼠NASH 中5-HETE含量下降,抑制铁死亡,缓解NASH。该研究指出,线粒体融合蛋白2(mitofusin 2,Mfn2)在砷暴露诱导的肝铁死亡上游发挥作用,同时Mfn2 可与肌醇需要酶1α (inositol-requiring enzyme 1 alpha,IRE1α)结合促进5-HETE 产生,引发脂质代谢紊乱,导致铁死亡发生、加剧NASH。因此Mfn2/IRE1α 可能通过ACSL4 正向调控铁死亡,加剧砷诱导的NASH。
综上所述,通过抑制脂质过氧化反应和铁死亡,可能是治疗NAFLD的可行方法。
1.4 NAFLD与铁死亡的其他诱发途径
线粒体氧化应激可能通过正向调控铁死亡参与NAFLD 的进展。线粒体氧化应激会导致其功能障碍,其特征是三羧酸循环流量以及线粒体ROS 增加,增强的线粒体ROS 可加速肝脂肪变性,补充辅酶Q10 可以逆转线粒体ROS 增加和肝脂肪变性[35]。膳食铁过载的情况下,黄鲶鱼肝脏中氧化应激增加,脂质过氧化物堆积,并出现了铁死亡。通过MitoTEMPO(线粒体超氧化物清除剂)处理显著降低黄鲶鱼肝细胞线粒体以及细胞质中的MDA 以及ROS,并增加Nrf2、GPX4 和GSH 等铁死亡中抗氧化酶的活性。之后通过溴化乙锭(ethidium bromide,EB)消融肝细胞中的线粒体,在铁过载的条件下消融线粒体,细胞ROS 并未增加,铁死亡受到抑制[39]。这说明,铁过载诱导的肝脏铁死亡是通过氧化应激导致的,且线粒体氧化应激占主要地位。 二氢乳清酸脱氢酶(dihydroorate Dehydrogenase,DHODH)是一种线粒体内膜酶,可降低线粒体氧化应激[40]。据报道,在GPX4 过表达或敲低的癌细胞中,抑制DHODH都可增强铁死亡诱导剂对其的作用[41]。在线粒体中,DHODH 与GPX4 平行发挥抗氧化作用,独立于胞质中的GPX4[40],这可能在NAFLD 中的铁死亡发挥作用。综上所述,线粒体氧化应激对于肝脏中的铁死亡发生十分重要,通过消除线粒体中的ROS来抑制铁死亡可能是防治NAFLD的新手段。
近几年的研究显示,Nrf2 通过调控下游铁死亡相关因子改善NAFLD中发挥重要的作用。
2 Nrf2在铁死亡调控NAFLD中的作用
转录因子Nrf2 是一种碱性亮氨酸拉链蛋白,是许多抗氧化蛋白的关键转录因子。正常状态下,Nrf2 与Kelch 样ECH 相关蛋白1(Kelch-like ECHassociated protein 1-nuclear factor,KEAP1)结合,通过泛素-蛋白酶体途径不断降解,当处于亲电和氧化应激状态时,KEAP1 上的半胱氨酸残基被修饰,引起KEAP1 的结构发生改变,破坏了KEAP1与Nrf2 的结合,抑制了Nrf2 降解,Nrf2 进入到细胞核中与小肌肉腱膜纤维肉瘤(small musculoaponeurotic fibrosarcoma,MAF)蛋白二聚化,促进下游细胞保护基因的转录[42]。
Nrf2 的下游因子已被报道在NAFLD 和铁死亡中起到关键作用[43],最新研究显示,激活Nrf2 可以抑制NAFLD 小鼠的铁死亡[3,9]。因此,本文重点讨论Nrf2、铁死亡与NAFLD 之间的联系,探讨激活Nrf2 抑制铁死亡影响NAFLD 的发生与发展(图2)。
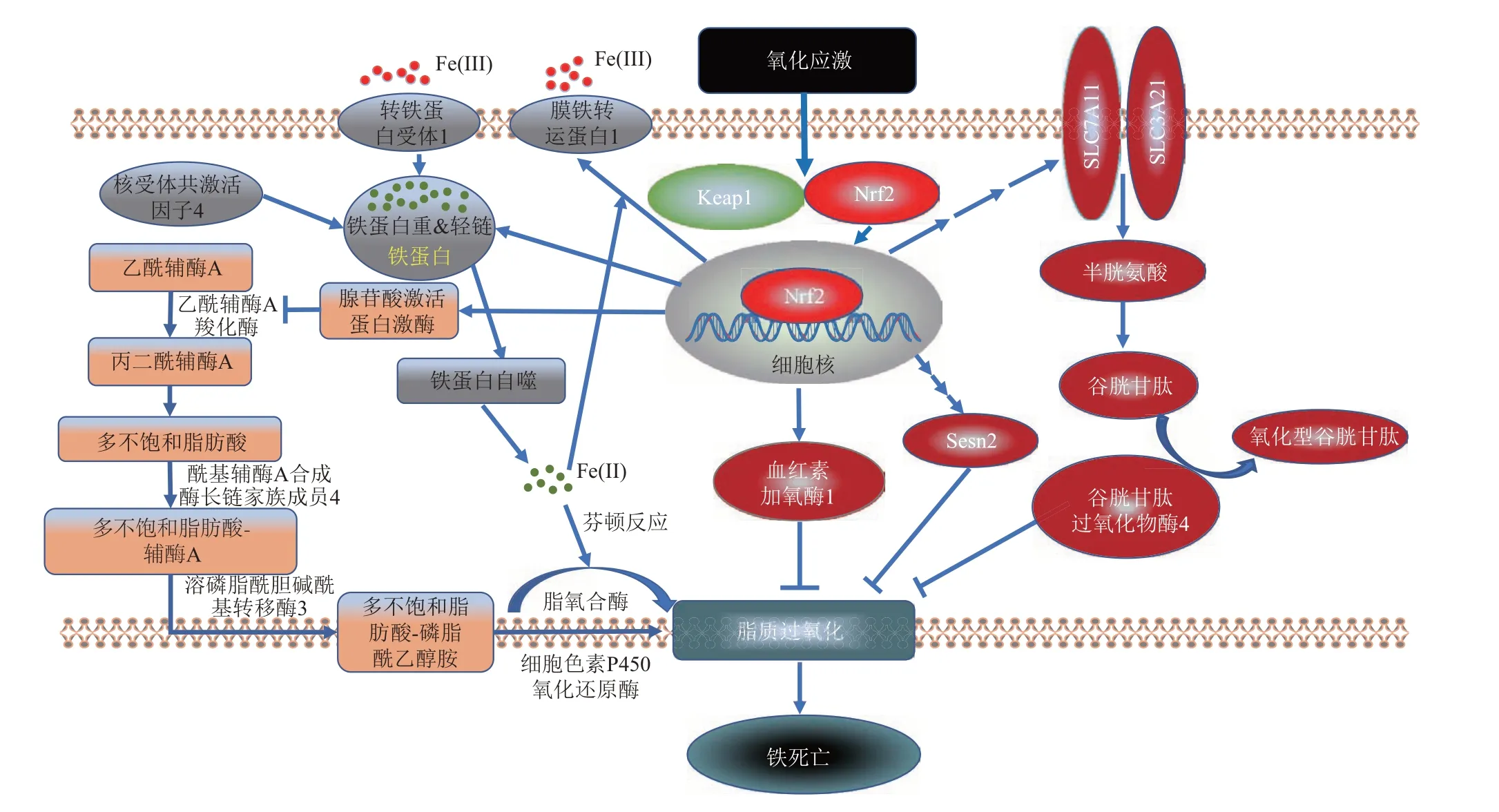
Fig. 2 The mechanism of ferroptosis regulated by Nrf2 in NAFLD图2 非酒精性脂肪肝中Nrf2调控铁死亡的机制
2.1 Nrf2与抗氧化
Nrf2 是抗氧化系统中的重要转录因子,可靶向一系列氧化还原相关基因,对铁死亡起到调控作用。
GPX4已被确立为Nrf2重要的抗氧化转录调节靶标[44],在多种细胞中沉默Nrf2 均可降低GPX4的表达[45-46]。除了直接调控GPX4转录外,Nrf2还可以通过调控GSH 的转录来调节GPX4 的活性。由上文可知,GPX4依靠GSH发挥抗氧化功能,因此GSH 是GPX4 的限速底物。由于参与GSH 的从头合成的胱氨酸/谷氨酸逆向转运体(system X C-,又名SLC7A11)、γ 谷氨酰半胱氨酸合成酶(γ-glutamyl cysteine synthetase,γ-GCS)和谷氨酸-半胱氨酸连接酶(glutamate-cysteine ligase,GCL)均受到Nrf2 调控。因此,Nrf2 是GSH 从头合成的重要的调控因子[47-48]。可通过调控GSH 合成间接影响GPX4的抗氧化功能。在结肠炎继发的肝损伤小鼠和四氯化碳诱导的急性肝损伤小鼠中,均可以通过激活Nrf2-GPX4 轴抑制肝脏铁死亡[49-50]。且在6 周的MCD 饮食后,与野生型小鼠相比,Nrf2敲除小鼠肝脏的脂肪变性、炎症和纤维化更为严重。同时Nrf2敲除小鼠肝脏中GSH水平明显下调、脂质过氧化物过度堆积[51]。这表明,Nrf2 可能直接或间接影响GPX4,从而抑制铁死亡改善NASH。除此之外,Nrf2还可以通过调控铁死亡抑制蛋白1(ferroptosis suppressor protein1,FSP1)的转录来调节抗氧化功能。FSP1 是一种新发现的内源性强铁死亡抑制剂,它执行氧化还原酶的作用通过减少辅酶Q10 增加铁死亡抗性[52]。在铁死亡中,FSP1与GPX4 平行发挥抗氧化功能[53]。有研究表明,在KEAP1 KO的肺癌细胞中,GPX4表达下降,而FSP1 表达增加,并表现出了铁死亡抗性。通过染色质免疫沉淀发现FSP1 的启动子中含有两个Nrf2的抗氧化反应原件(ARE)位点,在KEAP1 KO细胞中的Nrf2 缺失消除了KEAP1 KO 诱导的FSP1启动子-荧光素酶活性和FSP1 表达,并使KEAP1 KO 细胞对铁死亡诱导剂诱导的铁死亡重新敏感。这表明FSP1 是Nrf2 转录靶标,并且在GPX4 降低的情况下,Nrf2 可以通过上调FSP1 的转录发挥抗氧化功能,从而产生铁死亡抗性[54]。另外,HFD诱导的NAFLD 小鼠Nrf2 与FSP1 表达降低,ROS、MDA 和铁离子水平升高[3]。Nrf2 通过相互独立的GPX4 和FSP1 两种途径,协调发挥抗氧化、抗铁死亡的作用。这些可能为治疗NAFLD提供了一种启示。
除上述因子外Nrf2 还可通过影响HO-1[55]或Sestrin2(Sesn2)[56]发挥抗氧化功能。由上文可知,HO-1可产生胆绿素/胆红素和一氧化碳等代谢产物,这些代谢物可清除脂质过氧化物使细胞免受氧化应激的攻击[57]。目前多项实验证明通过增加NRF2/HO-1途径可以减缓NAFLD的发展[58-60]。一项研究发现,DA 通过与NRF2 竞争KEAP1,激活NRF2 下游HO-1 以及GSH 可以抗铁死亡,改善HFD 诱导的NAFLD[3]。提示,Nrf2 可通过激活HO-1 发挥抗氧化作用,抑制铁死亡改善NAFLD。Sesn2 是一种保守的抗氧化蛋白,响应于基因毒性、代谢和氧化应激等各种刺激,起到恢复机体平衡的作用[56]。研究表明,Nrf2可通过与Sesn2启动子中的ARE抗氧化反应原件结合刺激其转录[61-63]。Park 等[64]对原代肝细胞施加erastin(铁死亡诱导剂)诱导其发生铁死亡,发现Sesn2mRNA表达增加。为探讨在铁死亡中Sesn2升高是否由于转录调节,作者敲除Nrf2 后发现铁死亡诱导剂引起的Sesn2 升高消失。在删除Sesn2启动子上的ARE 反应原件后,Sesn2 表达下降。在铁过载诱导的肝损伤模型中过表达Sesn2 后,Sesn2 通过降低铁离子水平以及清除脂质过氧化物,抑制了铁死亡缓解肝损伤。利拉鲁肽(liraglutide,LG)可通过激活Sesn2、Nrf2/HO-1 改善肥胖诱导的NAFLD[65]。这些结果表明,Nrf2可能通过下游HO-1、Sesn2发挥抗氧化功能,抑制铁死亡改善NAFLD。
总而言之,Nrf2的抗氧化功能对于抑制铁死亡起到关键作用,它不仅可通过直接或间接调控GPX4活性,还可以在GPX4失活时刺激FSP1转录或直接刺激HO-1 和Sesn2 转录发挥抗氧化作用。因此,靶向Nrf2 可通过其抗氧化功能成为未来防治NAFLD的可行方法。
2.2 Nrf2与铁代谢
肝脏是铁储存的重要器官,Nrf2通过调控铁储存及转运控制铁代谢。铁积累增加导致铁代谢受损是铁死亡发生的原因之一。研究发现,在NASH模型中Nrf2 敲除小鼠体内铁积累比正常小鼠更加明显[51]。
肝脏中铁主要储存在铁蛋白中,铁蛋白重链(ferritin heavy chain, FTH1) 和铁蛋白轻链(ferritin light chain,FTL)是铁蛋白组成的一部分[66],FTH1含有一个亚铁氧化物酶活性位点,可以将Fe(II)氧化为Fe(II)储存在铁蛋白中,FTL可以控制铁蛋白的稳定[67]。FTH1和FTL都是已知的Nrf2 的转录靶点[68-69]。Nrf2 可以通过促进铁蛋白(FTL 和FTH1)的表达来调节铁的储存,以减少铁积累,从而减轻铁死亡[69]。在肝癌细胞中激活Nrf2 核易位可抑制肝癌细胞的铁死亡,沉默FTH1可增加肝癌细胞铁死亡[70]。在铁过载诱导的小鼠肝损伤模型中成纤维细胞生长因子21 (fibroblast growth factor 21,FGF21) 可激活Nrf2 及其下游靶基因GPX4、HO-1、FTH1 和FTL增加,通过增加抗氧化以及铁代谢功能抑制铁死亡,改善铁过载诱导的肝损伤[71]。银杏内酯B(ginkgolide B,GB)可激活Nrf2 通过抗氧化以及铁代谢抑制铁死亡,改善HFD诱导的NAFLD,但是在激活Nrf2后FTH1表达下调[9]。究其原因,一方面可能是由激活Nrf2 的物质(FGF21、GB)所决定,另一方面可能是由于铁蛋白自噬导致。上文了解铁蛋白自噬是维持机体铁稳态的重要机制[72]。NCOA4 作为自噬货物受体可以选择性地与FTH1相互作用,将其输送到溶酶体进行降解,最终导致FTH1 结合的铁释放,引起细胞内游离铁增加[73-74]。研究报道,铁蛋白在低铁条件下通过自噬降解,释放铁离子[75],因此HFD 可能导致铁蛋白自噬,FTH1 降低,当铁过载条件下,铁蛋白不发生自噬[75],FTH1为了降低铁过载,恢复机体铁稳态从而导致其表达增加。总之,Nrf2 可转录FTH1和FTL调控铁存储,改善肝脏中铁积累,抑制铁死亡,最终对NAFLD起到防治作用。
Nrf2不仅可以调控铁储存,还可以通过调控铁输出来调控铁代谢。FPN1 可将铁从细胞运输到血液循环中,以维持机体铁稳态[76]。FPN1的转录已被证明受Nrf2调控,Nrf2可通过调节FPN1的表达来影响细胞铁流量和细胞铁含量[77-78]。有研究表明,Nrf2/FPN1 通路通过介导铁稳态和铁死亡发挥改善疾病的作用[79]。并且女性NAFLD 患者中FPN1 增加[80]。因此,肝脏中Nrf2/FPN1 通路可能也会通过增加铁输出,抑制铁死亡达到改善NAFLD的作用。
总而言之,激活Nrf2 有望通过控制铁储存和铁输出降低铁过载引起的铁死亡,最终改善NAFLD。但目前研究多集中于Nrf2 影响铁储存改善NAFLD,而通过铁输出降低铁死亡改善NAFLD尚未见报道,需要后续研究证明。
2.3 Nrf2与脂质过氧化
抑制Nrf2 可以调控铁死亡过程中的脂质过氧化影响NAFLD。PUFA 既可由环境和饮食产生,还可通过(acetyl CoA carboxylase,ACC)产生,且ACC受Nrf2调控[81]。ACC是一种参与脂肪酸合成的酶,可催化乙酰辅酶A(acetyl-CoA)转化为丙二酰辅酶A(malonyl-CoA),而丙二酰辅酶 A是PUFA合成所必需的。研究表明,腺苷酸激活蛋白激酶(AMP-activated protein kinase,AMPK)可介导ACC 的磷酸化和失活负调节脂质过氧化反应。据报道,在AMPK 基因缺失的细胞中施加ACC 抑制剂可消除AMPK 失活引起的铁死亡[82]。另外,Nrf2 敲除小鼠的肝脏中AMPK 水平下降[83]。与之一致的,在Keap1敲除小鼠的肝脏中AMPK水平升高[84]。这说明肝脏AMPK 可能受到Nrf2 调控,且Nrf2 可能通过AMPK 间接参与脂质过氧化反应,调控铁死亡。
Nrf2 通过调控过氧化物酶体增殖物激活受体γ(peroxisome proliferator-activated receptor gamma,PPARγ)参与铁死亡过程。PPARγ对脂质代谢至关重要[85-86],且PPARγ 由Nrf2 转录[87]。铁死亡中除ACSL4外,环氧合酶2(cyclooxygenase-2,COX2)同样可催化AA生成脂质过氧化物。在神经元细胞中铁死亡诱导剂可降低PPARγ表达,升高COX2的表达,激活PPARγ后可通过抑制COX2的表达,减弱脂质过氧化物生成,抑制铁死亡[88]。在原代神经元细胞中PPARγ 激活剂增加Nrf2 与PPARγ 的表达,抑制铁死亡诱导剂诱导的铁死亡[89]。且在HFD诱导的NAFLD小鼠中,Nrf2敲除小鼠与正常小鼠相比,脂质堆积减少,PPARγ 水平降低[90]。这说明,受Nrf2 所调控的PPARγ 可能通过参与脂质过氧化反应调控铁死亡,影响NAFLD的进展。
综上所述,Nrf2 有望通过直接影响PPARγ 或通过节间影响AMPK,调控脂质过氧化反应,抑制铁死亡。
2.4 Nrf2其他机制
上文可知,线粒体功能紊乱参与NAFLD 与铁死亡中。在线粒体功能紊乱的疾病中,Nrf2 也受到抑制[91]。据报道,Nrf2 敲除小鼠的线粒体ROS明显高于WT 小鼠[92]。而且,线粒体是细胞ROS的主要来源,同时线粒体也是产生腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)的主要细胞器。然而,当线粒体障碍能量代谢受损时,会激活受Nrf2 调控的AMPK,它通过限制ACC 参与脂质过氧化反应,调控铁死亡。且AMPK 失活会激活铁死亡[82]。萝卜硫素(glucoraphanin)[93]或一氧化碳释放分子A1 (Carbon monoxide releasing molecule-A1,CORM-A1)[94]可激活Nrf2通过影响线粒体功能障碍改善NAFLD。这提示在NAFLD中激活Nrf2 可能通过调节线粒体功能障碍抑制铁死亡改善NAFLD。
3 结论与展望
NAFLD 因其高发病率和潜在的危害受到广泛关注,并与铁死亡高度相关[19]。本文讨论了铁死亡在NAFLD 中的机制,铁死亡通过GPX4 途径、铁代谢途径、脂质代谢途径以及一些其他途径参与到NAFLD 的进程中。此外,多项研究表明Nrf2、铁死亡和NAFLD存在一定联系,Nrf2通过抑制铁死亡可改善NAFLD[10-11]。
针对目前铁死亡改善NAFLD 的研究,本文提出以下几点思考。在NAFLD 的进展中,GPX4 的表达在NAFLD 与NASH 中出现相反的结果,本文猜测这可能是机体对于升高炎症或脂质过氧化反应做出的应激反应。而FSP1 与GPX4 均通过抗氧化功能,达到抵抗铁死亡的目的。但FSP1在NAFLD中的研究尚浅。GPX4 与FSP1 可能均受到Nrf2 的调控。探讨在NAFLD 中靶向Nrf2、GPX4 与FSP1分别有什么影响,以及是否可同时激活这两种抗氧化途径,以在NAFLD 中共同发挥抑制铁死亡的功能,具有重要的意义。
由于肝脏在铁储存中的重要性,铁过载又是NAFLD 与铁死亡发生的重要原因,了解何种途径降低铁过载,抑制铁死亡,可能会对NAFLD 的防治具有极大的帮助。但由于肝脏铁代谢的复杂性,目前研究尚不全面,仍需进一步的探索。例如,肝脏线粒体中铁代谢是否参与NAFLD 的铁死亡,铁过载与脂质过氧化的复杂关系,Nrf2 调控的FTH1在铁过载以及HFD 小鼠中的表达,受Nrf2 调控的FPN1 作为人体内唯一的铁输出蛋白,在NAFLD中也起到调控铁代谢功能,但是FPN1如何通过调节铁死亡从而调控NAFLD,对于上述问题的研究均未见报道。
另外,目前研究较少将铁死亡中脂质过氧化途径的一些重要酶与NAFLD 建立关联,如将游离不饱和脂肪酸转移到磷脂中的LPCAT3及合成脂质过氧化物的关键酶等。因此,需要后续研究证明这些参与脂质过氧化反应的因子是否可影响NAFLD 的进程。
总体而言,通过Nrf2 抑制铁死亡改善NAFLD是可行的。望后续发现更多与Nrf2 相关的途径,为改善NAFLD提供帮助。
