快速制备水溶性RAFT聚合链转移剂的方法研究
2023-08-10杜思思杨永启伊祖江冯国瑞许振广李煜闫友军
杜思思,杨永启,伊祖江,冯国瑞,许振广,李煜,闫友军
(潍坊科技学院 山东省化工资源清洁利用工程实验室 潍坊市农业秸秆绿色高值化利用重点实验室,山东 潍坊 262700)
可逆加成-断裂链转移自由基聚合(RAFT)是1998年由澳大利亚三位科学家Graeme Moad,Ezio Rizzardo和San H.Thang提出的一种活性聚合方法[1-2]。RAFT聚合机理通常认为经历下面五个过程[3-4],即:1)链引发阶段;2)链增长阶段;3)增长链自由基形成阶段;4)活性休眠种形成阶段;5)链终止阶段。RAFT聚合与其他活性聚合相比优势在于,特异性的选择链转移剂二硫代或三硫代碳酸酯,可以通过调控活性休眠种达到调控聚合反应速率的目的。RAFT聚合的一大优势就是活性链的保持性,得到的聚合物因端基仍然保留高活性的二硫代酯或三硫代酯,加入单体后活性链还可以继续增长,可以制备控制性很好的多嵌段和超高分子量聚合物,这是其他活性自由基聚合方法(如NMP和ATRP)无法企及的[5-6]。RAFT聚合的另一大优势是聚合单体种类范围广。RAFT聚合之所以能够适应单体种类广,很重要的一点是在自由基加成反应过程中RAFT试剂的C=S键比单体的C=C键活泼,因此选择合适的离去基团(R基团)和合适的稳定基团(Z基团)对于RAFT试剂来说至关重要。Z基团在自由基加成和稳定自由基中间体方面起重要作用,R基团的选择必须同时考虑自由基的稳定性和空间效应,必须满足R基团能够形成稳定的自由基,同时还需满足R基自由基能够很好地与单体反应形成增长链自由基[7-8]。
虽然已知文献报道的RAFT试剂种类比较繁多,但是普遍存在的一个问题是这些RAFT链转移剂含有长链的憎水基团,通常在水溶液中溶解性很差,这大大限制水溶液中RAFT聚合反应的进行,如何提高RAFT链转移剂在水中溶解度一直是广大科研工作者们研究的热点和难点[9]。本文经过反复实验探究,得出一种快速制备水溶性RAFT聚合链转移剂的制备方法。
1 实验部分
1.1 实验原料及仪器设备
实验原料及主要仪器设备见表1和表2。
表1 实验原料及厂家信息

表2 主要仪器设备及厂家信息
1.2 制备过程
1.2.1 4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸
其方法及步骤如下:
1)在1 000 mL圆底烧瓶中,室温下依次加入20 g乙硫醇和500 mL乙醚,混合搅拌18 min 后分四次加入14 g氢氧化钠固体,继续搅拌反应2.5 h后小心逐滴滴入26.70 g CS2,溶液颜色将快速由无色变为黄色并很快出现黄色浑浊物,继续搅拌反应8 h后将黄色浑浊液过滤,收集黄色固体粗产物,固体粗产物用回收旋蒸纯化后的乙醚反复洗涤三次得到质量为30.12 g,此即为三硫代碳酸钠盐粗产物;
2)准确称取11 g三硫代碳酸钠盐粗产物溶于160 mL乙醚中搅拌形成均匀的悬浊液,加入9.63 g碘单质,反应2 h后停止搅拌;然后准备一个500 mL的烧杯,向其中加入上述反应液,额外加入160 mL水混合后倒入1 L分液漏斗中,用分液漏斗混合分液,随后加入11 g适量硫代硫酸钠除去过量的碘单质,此时水层颜色逐渐变浅,收集乙醚层混合物,继续加水130 mL,重复洗涤3次,收集上层乙醚液混合物,加入无水氯化钙干燥3 h后过滤,蒸馏除去溶剂得到六硫代化合物;
3)将5.7 g六硫代化合物和155 mL乙酸乙酯加入500 mL 双口圆底烧瓶中,加入9.14 g ACVA,冰水浴搅拌,装球形冷凝管密闭后鼓氮气除氧气40 min,继续通氮气将反应瓶转移到预热好的80 ℃油浴锅中搅拌下回流反应8 h;然后准备一个1 L的烧杯,向其中倒入上述反应液,搅拌条件下再加入体积为40 mL的饱和NaCl溶液,倒入体积为1 L的分液漏斗中混合后萃取分液,收集乙酸乙酯层,重复萃取5次后收集上层液体,倒入密闭圆底烧瓶中,加入一定量无水氯化钙除水干燥,静置3 h后抽滤收集滤液,通过旋转蒸发仪在50 ℃水浴加热下真空旋蒸除去萃取后的溶剂1 h后得到颜色为橘红色的液体,然后在玻璃柱中填装45~75 μm(200~300目)硅胶粉,将上述黏稠的橘红色液体过柱,洗脱剂选择乙酸乙酯和石油醚体系,先用体积比为5%的乙酸乙酯/石油醚混合溶液冲柱分离杂质,然后选择10%乙酸乙酯/石油醚混合溶液冲出杂质,最后用当体积比为30%的乙酸乙酯/石油醚混合溶液分离得到纯净的小分子RAFT链转移剂4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸(CTA-1,8.97 g,产率82%)。制备反应历程见图1。

图1 CTA-1的制备反应历程
1.2.2 2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯
当流完后提高洗脱剂极性至V(乙酸乙酯)∶V(石油醚)=70∶30时得到尚未反应的六硫代化合物,回收待用;在100 mL圆底烧瓶中依次加入0.906 9 g的 CTA-1,1.326 2 g PEG8-OH,0.084 9 g 的4-二甲氨基吡啶和30 mL 二氯甲烷。搅拌15 min后用注射泵将溶于10 mL 二氯甲烷的0.922 8 g 二环己基碳二亚胺逐滴加入上述混合液中,继续室温搅拌反应12 h。反应结束后将混合体系通过砂芯漏斗除去白色沉淀物,收集滤液,先后用水(4×110 mL)和100 mL的饱和NaCl溶液洗涤滤液,收集二氯甲烷层液体加入无水氯化钙干燥3 h后,通过旋转蒸发仪50 ℃水浴真空旋蒸除去上述混合溶剂,得到颜色为黄色的黏稠状液体,在45~75 μm(200~300目)硅胶柱中洗脱分离杂质,旋蒸甲醇与二氯甲烷体系做洗脱剂,先用纯净的二氯甲烷冲洗体系,然后选择体积比1%的甲醇/二氯甲烷混合溶液冲出杂质,最后用体积比为2%的甲醇/二氯甲烷混合溶液冲出得到目标产物,通过旋转蒸发仪在50 ℃水浴加热下真空旋蒸除去溶剂后得到水溶性RAFT链转移剂2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯(PEG8-CTA,1.65 g,产率87%)。其制备反应方程式如图2所示。

图2 PEG8-CTA的反应方程式
通过上述反应过程可知,反应过程转化率较高,总结其制备工艺流程图如图3所示。

图3 水溶性链转移剂PEG8-CTA的制备工艺流程图
2 实验结果及分析
2.1 4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸的核磁共振表征
取15 mg 4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸液体溶于500 μL的氘代氯仿中,在400 M核磁共振波谱仪上测试扫描磁场16次,得到其核磁共振氢谱图谱如图4。
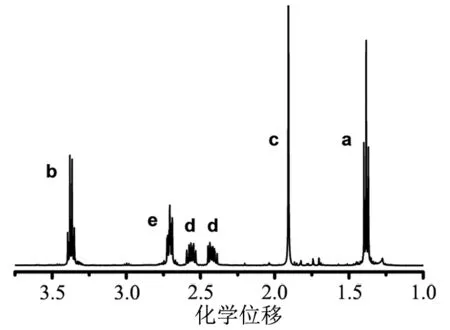
图4 CTA-1的核磁共振氢谱谱图(CDCl3中)
由图4可知,CTA-1的核磁共振氢谱已经纯化,残留的少量溶剂可以通过旋转蒸发仪去掉,不影响后续反应,其核磁共振氢谱数据及位置情况分析如下:1H NMR(400 MHz,CDCl3)δ:1.3 ppm(t,3H,-CH3),1.9 ppm(s,3H,-CH3),2.4~2.6 ppm(t,2H,-CH2-),2.7 ppm(t,2H,-CH2-),3.4 ppm(q,2H,-CH2-)。
2.2 2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯的核磁共振氢谱表征
取15 mg 2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯液体溶于500 μL的氘代重水中,在400 M核磁共振波谱仪上测试扫描磁场16次,得到其核磁共振氢谱图谱如图5。
由图5可知,PEG8-CTA的核磁共振氢谱已经纯化,其纯度大于98%,完全达到分析纯纯度要求,其核磁共振氢谱数据及位置情况分析如下:1H NMR(400 MHz,D2O):δppm 1.23(t,3H),1.77(s,3H),2.43(t,2H),2.56(t,2H),3.24(s,3H),3.27(t,2H),3.55(m,30 H),4.11(t,2H)。
2.3 2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯的核磁共振碳谱表征
取40 mg 2-[(2-甲氧基乙氧基)乙基]-4-氰基-4-[乙硫基(硫代羰基)硫代]戊酸酯液体溶于500 μL的氘代氯仿中,在400 M核磁共振波谱仪上测试扫描磁场512次,得到其核磁共振碳谱图谱如图6。

图6 具有良好水溶性的RAFT链转移剂PEG8-CTA的核磁共振碳谱图(氘代氯仿中)
由图6可知,PEG8-CTA的核磁共振碳谱已经纯化,其纯度大于98%,完全达到分析纯纯度要求,其核磁共振碳谱数据及位置情况分析如下:13C NMR(400 MHz,CDCl3):δppm 12.82,24.82,29.70,31.38,33.76,46.35,59.05,64.18,68.97,70.61,71.93,119.03,171.49,216.84。
3 结论
报道了一种制备工艺简单、原料易得且水溶性良好的RAFT链转移剂的制备的方法,所有的制备原料便宜易得,装置简单,大大节约成本,纯化过程涉及萃取、过柱和蒸馏操作,操作简单,不需要复杂设备;制备过程中除制备CTA-1过程需要80 ℃高温外,其余均可以室温进行,可以降低生产成本;所有的溶剂二氯甲烷、石油醚、甲醇、乙酸乙酯都可以回收循环使用,节约成本;探究的实验比例重现性好,纯度高,可以放大中试生产,方便工业大规模制备。
