长枝木霉菌非核糖体肽合成酶基因启动子NP249的克隆及功能验证
2023-07-08官萌娇孙旭杰夏卓林任爱芝赵培宝
官萌娇,孙旭杰,夏卓林,任爱芝,赵培宝
(聊城大学 农学与农业工程学院,山东 聊城 252000)
木霉菌(Trichodermaspp.)属半知菌亚门,多数菌株进行无性繁殖,有性阶段为肉座菌,是重要的生防真菌。对环境适应性强,生防机制多样,已被广泛应用于防治由真菌、细菌和线虫引起的植物病害[1-2]。木霉菌对植物也有一定的促生作用,能够提高植物对逆境的适应能力,最终促进植物的生长。木霉产生的次级代谢产物及抗生素在生防过程中起着重要的作用。
木霉菌次生代谢产物多样,Peptaibols是真菌的次生代谢产物之一,是由非核糖体多肽合成酶(Nonribosomal peptide synthetase,NRPS)合成的多肽类抗菌物质,具有抗病毒、抗真菌和抗癌等多种生物学活性[3-6]。长枝木霉SMF2(TrichodermaLongibrachiatumSMF2)为一株高产Peptaibols的菌株,该菌株能够产生Peptaibols类抗菌素康宁霉素(Trichokonins,TKs),其中Trichokonin VI含量最高[7]。Trichokonin VI能够抑制革兰氏阳性细菌枯草芽孢杆菌的生长[8],对臭椿苗期病害以及灰葡萄孢菌引起的蝴蝶兰灰霉病也有一定的防治效果[9-10];激活水杨酸信号途径,增加大白菜对软腐病的抗性[11];通过激活多条防御信号通路增加烟草对烟草花叶病毒侵染的防御反应和系统抗性,诱导烟草体内ROS的产生、施药部位酚类化合物的积累和对病毒的抗性[12]。由此可见,Trichokonin VI在生物防治方面具有广阔的应用前景,但其合成机制尚不清楚,因此,研究其合成机制,提高Trichokonin VI产量使其在生物防治中发挥作用是至关重要的。随着长枝木霉SMF2基因组序列的公布,长枝木霉SMF2合成Peptaibols机制的研究揭开了新篇章,除了添加特定氨基酸和调控培养基的碳氮源配比等条件来调控Peptaibols的产生,Peptaibols的生物合成还受到多种基因的调控。Peptaibols是一类由非核糖体合成酶系统合成的多肽,其氨基酸序列分别与NRPS上的各个组件相对应,这就为Peptaibols的分子改造提供了可能[13]。通过对长枝木霉菌 SMF2 菌株产生的抗菌素进行深入研究,明确了该菌株产生的Peptaibols短肽TrichokoninVI,由 NRPS 基因簇TPX1合成[14]。
为进一步探究非核糖体肽类抗菌素的合成机制,本研究以长枝木霉SMF2菌株为材料,克隆了NRPS的启动区域,构建绿色荧光表达载体,采用REMI介导的原生质体转化法,将重组的携带GFP基因的质粒pCX-62-GFP-249转入长枝木霉SMF2野生型菌株中,验证启动子功能。
1 材料和方法
1.1 试验材料
1.1.1 菌株与引物 长枝木霉SMF2由聊城大学农学院赵培宝实验室保存;绿色荧光蛋白GFP基因来自pCX-62-GFP由本实验室前期构建;引物249-1:5′-ATGTGGGAAATAGCGAGAT-3′(60);249-4:5′-GGGCTGCCAGGTCTTTT-3′(NRPS22);249-1B:5′-CGGGATCCATGTGGGAAATAGCGAGTA-3′;249-4X:5′-TCCCCCCGGGCTTGGCGGGTTGGGCG-3′,由上海生工合成。
1.1.2 主要试剂 琼脂糖凝胶DNA回收试剂盒、高纯质粒小量制备试剂盒、真菌基因组DNA提取试剂盒百泰克(BioTeke)公司;T4DNA连接酶、XmaⅠ、BamH Ⅰ、pMD-18T载体等购自宝生物(TaKaRa)公司;其他试剂均为国产分析纯。
1.2 试验方法
1.2.1 基因组DNA的提取 将长枝木霉转接到PDA培养基上培养5 d,刮取菌丝烘干。烘干后加入液氮研磨,使用真菌基因组DNA试剂盒提取DNA,-20 ℃保存备用。
1.2.2 启动子预测 采用Promoter 2.0(http://www.cbs.dtu.dk/services/Promoter/)、TFSEARCH(http://www.Gene-regulation.com)等在线软件,参考EPD(Eukaryotic promoter database,http://www.epd.Isb-sib.ch/)、TRANSFAC(http://www.Gene-regulation.com)等启动子数据库,并结合真菌启动子的结构特征,对NRPS基因TPX1上游DNA序列进行分析,寻找其顺式元件,例如TATA盒、CAAT盒、GC盒及其他可能的调控元件,并结合荧光蛋白GFP等报告基因的融合与表达分析来确定启动子。
1.2.3 启动区扩增 以长枝木霉SMF2的DNA为模板,249-1和249-4为引物进行PCR扩增,95 ℃预变性5 min;94 ℃变性45 s,56 ℃退火45 s,72 ℃延伸1 min,30个循环;72 ℃最终延伸10 min。得到1 204 bp的目的基因249的片段,送去测序。
测序后的片段与pCX-62所含的酶切位点相比较分析,在249-1和249-4的5′端添加酶切位点和保护碱基设计带有酶切位点的引物249-1B和249-4X。PCR扩增以扩增出的片段为模板,249-1B和249-4X为引物进行PCR扩增,95 ℃预变性5 min;94 ℃变性45 s,68 ℃退火45 s,72 ℃延伸1 min,30个循环;72 ℃最终延伸10 min。
胶回收纯化后得到NP249基因启动子片段,将目的片段DNA连接到pMD-18T载体上,在PCR仪内过夜连接得到重组载体NP249-pMD-18T,通过大肠杆菌热激法转化将重组质粒249-pMD-18T转化至DH5α菌体内实现重组质粒的扩增。
1.2.4 GFP融合载体构建 用XmaⅠ、BamH Ⅰ对pCX-62-GFP和249-pMD-18T进行酶切,T4DNA连接酶16 ℃过夜连接,将连接后的质粒通过大肠杆菌热激转化法导入到大肠杆菌DH5α感受态细胞内。以菌液为模板,249-1B和249-4X为引物,进行PCR扩增验证菌液是否为阳性,菌液PCR结果为阳性后提取重组质粒249-pCX-62-GFP双酶切鉴定。
1.2.5 遗传转化和转化子筛选 制备长枝木霉SMF2原生质体,保存的长枝木霉菌SMF2转接于PDA上,28 ℃培养3~5 d后刮取孢子,置于50 mL PDB培养基中,28 ℃,150 r/min振荡培养12 h。等量分装于25 mL离心管中,放入高速冷冻离心机中,4 000 r/min,4 ℃离心10 min,收集菌丝。加入10 mL 0.7 mol/L NaCl溶液冲洗菌丝,4 000 r/min,4 ℃离心10 min,弃上清液,后重复此步骤1次。称取0.1 g溶壁酶与4 mL 0.7 mol/L NaCl溶液混合,混匀后用0.22 μm水相针式滤器过滤,过滤后的溶液加入收集菌丝的离心管内,混合均匀,置于恒温振荡培养箱中30 ℃,150 r/min培养3.5 h得到原生质体。制备好的原生质体中加入28 mL 0.7 mol/L的NaCl溶液,用3层擦镜纸过滤至新的离心管中,4 000 r/min,4 ℃离心7 min,收集原生质体。
使用限制性内切酶介导的转化(REMI)技术对长枝木霉SMF2原生质体进行转化,在含原生质体的离心管内加入10 mL STC溶液冲洗离心弃掉上清液,加入150 μL STC备用。将50 μL STC溶液、50 μL线性化质粒、2 μL限制性内切酶Hind Ⅲ加入1.5 mL离心管中,混合均匀后加入上述含150 μL STC的原生质体中,冰浴20 min。加入2 mL预冷的PTC,冰浴20 min,室温20 min。加入20 mL STC,4 000 r/min,4 ℃离心7 min,弃掉上清液,加入2 mL再生PDB置于28 ℃过夜培养,将过夜培养的长枝木霉原生质体与再生PDA混合均匀倒平板,待长出微菌落后覆上10 mL含100 μg/mL潮霉素的0.7%水琼脂,28 ℃培养。
1.2.6 转化子筛选 待长出转化子后将长出的转化子转接到含潮霉素抗性的培养基上,连续转接3代筛选稳定的转化子,提取转化子DNA,以Hph1和Hph2为引物进行PCR验证。挑取转化子的菌丝置于荧光显微镜下检测荧光表达状态,验证启动子功能。
2 结果与分析
2.1 启动子预测
通过网站获取TPX1基因转录起始点上游1 204 bp作为启动子序列,根据启动子预测结果(图1),可见大号的A是酶转录起始碱基,上游30 bp处有一个TATA box。

图1 启动子预测Fig.1 Promoter predictions
2.2 启动区的扩增
以长枝木霉SMF2基因组DNA为模板进行PCR扩增,扩增产物回收测序结果显示成功克隆NP249启动子序列(图2)。将测序正确的片段连接到pMD-18T载体上,提取质粒,经1%琼脂糖凝胶电泳分析可见目的基因条带,大小与预期一致(图3)。

M.DL2000 DNA Marker;1,2.NP249基因片段。M.DL2000 DNA Marker;1,2.NP249 gene fragment.

M.DL5000 DNA Marker;1.pMD-18T重组质粒。M.DL5000 DNA Marker;1.pMD-18T recombinant plasmid.
2.3 融合载体的构建
连接过夜的质粒转入大肠杆菌DH5α感受态细胞内,菌液PCR验证为阳性(图4)。249-pCX-62-GFP的双酶切(XmaⅠ、BamH Ⅰ)产物经1%琼脂糖凝胶电泳可见2条条带,载体条带和目的基因条带,大小与预期一致,表明获得了249启动子的GFP融合载体(图5)。

M.DL2000 DNA Marker;1.目的片段;2.GFP融合载体菌液PCR。M.DL2000 DNA Marker;1.Target fragment;2.GFP fusion vector bacterial solution PCR.

M.DL5000 DNA Marker;1.GFP融合载体双酶切。M.DL5000 DNA Marker;1.Double enzyme digestion of GFP fusion vector.
2.4 转化子的荧光检测
提取转化子DNA,进行PCR扩增,扩增得到潮霉素抗性条带(图6),表明载体已整合到基因组中。使用荧光显微镜对转化子菌丝进行荧光检测,检测到GFP荧光信号(图7),表明克隆到的基因启动子能够启动GFP的表达,具有启动子功能。
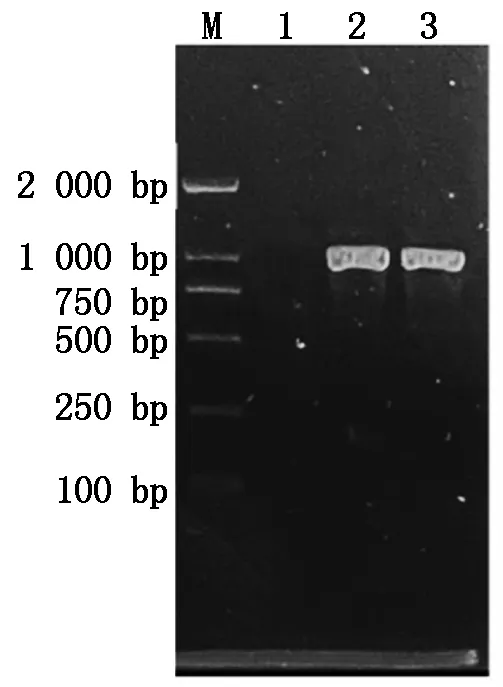
M.DL2000 DNA Marker;1.野生型长枝木霉SMF2 DNA;2,3.转化子DNA。M.DL2000 DNA Marker;1.DNA of Trichoderma longibrachiatum SMF2;2,3.DNA of transformants.
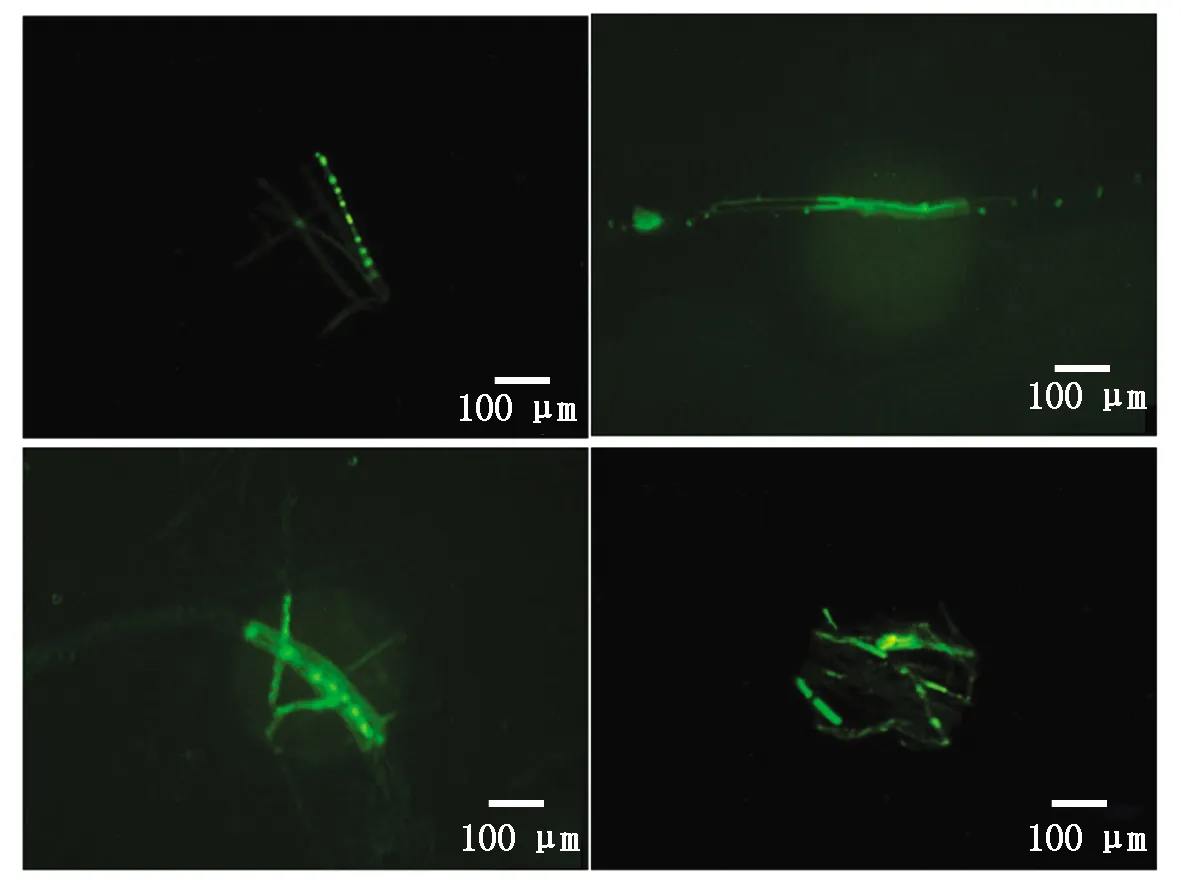
图7 转化子菌丝荧光检测Fig.7 Fluorescent detection of transformant hypha
3 结论与讨论
综上所述,本试验扩增到的1 204 bp大小的NRPS启动子序列能够启动GFP的表达,具有启动子的功能。该启动子的克隆为进一步研究NRPS合成Peptaibols的机制奠定了基础。
Peptaibols是木霉最主要的次级代谢产物之一,是由NRPS催化单个氨基酸残基添加至肽链合成的非核糖体肽类。非核糖体合成酶合成多肽过程遵循共线性装配规则,非核糖体合成酶途径通过利用除20种氨基酸外的稀有氨基酸或脂肪酸为原料,在多酶或多结构域合成系统的参与下合成非核糖体肽(Nonribosomal Peptides,NRPs),合成机理为多载体巯基化模板[15]。
NRPSs是一种分子量巨大的多功能酶系,是合成NRPs的关键。它由一系列模块按顺序排列作为多肽合成的模板,因而模块的种类、数量和空间排列顺序决定了多肽的特异性,且模块在基因组上成簇排列。每一个模块由负责识别并活化特定底物为相应的腺苷酸化合物的腺苷酸化结构域(Adenylation,A)、起反应中间物硫脂固定作用的巯基化结构域(Thiolation,T)和负责肽键形成的缩合结构域(Condensation,C)共同组成,从而实现将一种氨基酸整合至产物骨架中[16-20]。巩志廷[21]发现,在长枝木霉SMF2中,TPX1和TPX2基因分别控制含有20个氨基酸残基和12 个氨基酸残基的Peptaibols的生物合成;Zhou等[22]通过敲除TISTP1基因使Trichokonin A的产量增加了5倍,Trichokonin B的产量增加了2.6倍;Shi等[23]发现敲除偏甲基转移酶基因TLIae1下调了Tlx1和Tlx2的表达,抑制了Trichokonin A和Trichokonin B的产生。生物信息学分析发现,在长枝木霉SMF2中有NRPSs的同源基因,该基因可能与NRPSs合成Peptaibols相关,但对其如何发挥作用还不清楚。
通过对启动子进行克隆明确该基因具有启动子功能,继而寻找反式作用因子,来研究NRPS基因表达调控方式,为NRPS转录调控,高水平合成非核糖体抗菌肽提供帮助。
