ST37型艰难梭菌分子流行病学研究
2023-04-29陈熔吕晓菊
陈熔 吕晓菊
摘要:目的 應用全基因组分析方法对艰难梭菌感染可能的暴发流行进行识别和调查,为艰难梭菌感染防控提供可靠的分子流行病学基础。方法 对8株ST37型艰难梭菌进行第二代高通量测序,并完成序列的拼接和注释。通过对其核心基因组进行SNP分析,根据SNP数量以及相关临床资料分析有无院内暴发流行。同时将Genbank中公布了全基因组序列的艰难梭菌,进行MLST分析,对其分析结果为ST37的菌株与本研究中的8株菌株进行SNP分析比较,了解这8株菌的可能来源。结果 以最早收集的菌株WCHCD770作为参照,其他7株菌株的SNP值,最大为59,最小为38,提示它们并不是近期发生的传播事件。而这8株菌两两相互进行SNP计算,其中WCHCD1577、WCHCD1641仅为10,提示它们可能来源于一个克隆,可能存在院内传播。通过分析比较这8株菌与Genbank中的其他ST37型艰难梭菌发现,它们的致病决定区(PaLoc)序列完全相同,证实了ST37型艰难梭菌的PaLoc在世界范围克隆传播,同时对它们的SNP比较分析,发现WCHCD1577、WCHCD1641、WCHCD1216、WCHCD109、WCHCD159与2006年在爱尔兰分离的菌株M68的SNP最小,WCHCD770、WCHCD1262、WCHCD1450与加拿大分离的菌株VL-0005的SNP最小。提示这些菌株的可能来源。结论 8株ST37型艰难梭菌可能存在克隆传播,值得感染防控重视。
关键词:艰难梭菌;抗生素相关性腹泻;多位点序列分型;单核苷酸多态性
中图分类号:R978文献标志码:A
Abstract Objective To identify and investigate the outbreak of Clostridium difficile infection using whole genome sequence, and provide basic molecular epidemiology for preventing and controlling Clostridium difficile ST37. Methods High-throughput genome sequencing and annotation of eight clinical isolates of Clostridium difficile ST37 were completed. Single nucleotide polymorphism was analyzed by the core genome. Whether there is nosocomial infection outbreak was analyzed according to the number of SNPs. The whole genome sequence of Clostridium difficile was deposited in Genbank, and multiple locus sequence typing (MLST) was conducted and analyzed. SNP analysis of ST37 strains and our eight clinical isolates were performed in order to understand the possible source of our eight clinical isolates. Results Using the earliest strain WCHCD770 as the reference, we found the biggest SNPs was 59, the smallest one was 38. It suggested that they were not recent spread events. But the smallest SNP of the eight clinical Clostridium difficile strains was 10, which was between WCHCD1577 and WCHCD1641, indicating possible nosocomial transmission. Comparing to other ST37 strains in Genbank, we found Pathogenicity Locus (PaLoc) sequences of all Clostridium difficile ST37 were identical. It was confirmed that PaLoc of Clostridium difficile ST37 was spread around the world. We found the Clostridium difficile strain M68 which was collected from Ireland in 2006 had smaller SNP than WCHCD1216, WCHCD1577, WCHCD1641, WCHCD109, and WCHCD159. At the same time, the strain VL-0005 which was collected from Canada had smaller SNP than WCHCD770, WCHCD1262, and WCHCD1450. This suggested possible strains source. Conclusion This experiment confirmed the possible spread event and source in Clostridium difficile ST37, which was helpful to guide prevention and control of Clostridium difficile.
Key words Clostridium difficile; Antibiotic-associated diarrhea; MLST; SNP
艰难梭菌是引起抗生素相关性腹泻(antibiotic-associated diarrhea, AAD)和伪膜性肠炎(pseudomembranous colitis, PMC)的重要致病菌[1-3]。自2001年以来,艰难梭菌感染(Clostridium difficile infection, CDI)在全球呈上升趋势,其高毒力菌株PCR-核糖体分型(RT027)感染在北美和欧洲爆发流行[4-5]。亚洲国家对艰难梭菌的研究相对较少,RT027引起的CDI只有零星的报道。相关报道中,RT017(MLST分型属于ST37)为亚洲主要流行型别[6-7]。而中国大陆目前只有部分大城市医院有检测和研究报道[8]。本研究从临床患者分离获得8株ST37型艰难梭菌,由于菌株短时间内来源于同一病房,它们之间是否存在院内传播积极探索。
1 材料与方法
1.1 材料
1.1.1 菌株来源
收集四川大学华西医院ICU病房2014年8月—12月临床腹泻患者大便标本。腹泻的定义为24 h内出现3次或以上的腹泻。
1.1.2 仪器与试剂
台式离心机(德国Eppendorf公司),基因扩增仪、电泳仪(美国Bio-Rad公司),脑心浸出液(BHI)肉汤(青岛高科园海博生物技术有限公司),艰难梭菌选择培养基CCFA、厌氧产气袋(英国Oxoid公司),细菌DNA提取试剂盒(美国Omega公司),SPAdes(序列拼接软件),Prokka(基因注释程序),Jmodeltest、PhyML tools(SNP分析软件)。
1.2 方法
1.2.1 粪便厌氧培养及鉴定
粪便标本先用75%酒精处理,再接种于CCFA琼脂平板,放置于塑料密封袋中,再加入厌氧产气袋,厌氧指示剂,夹上密封夹,置于培养箱37℃孵育48 h。挑取可疑单个菌落传代接种于CCFA培养基上,厌氧培养48 h后取单个菌落进行涂片、革兰染色,置显微镜下观察,经生化鉴定为艰难梭菌后,进一步采用PCR技术检测毒素基因(tcdA和tcdB)。选取7个管家基因(adk, atpA, dxr, glyA, recA, sodA和tpi)进行PCR扩增并测序,将序列与数据库比对,分析菌株的ST型别。
1.2.2 基因DNA的提取
ST37型菌株根据细菌DNA提取试剂盒说明书操作,提取细菌基因组DNA。
1.2.3 全基因组测序
将提取的艰难梭菌染色体DNA送诺和基因公司进行二代测序,测序数据使用SPAdes和Prokka软件进行拼接和注释。
1.2.4 数据分析
使用Jmodeltest和PhyML Tools软件对收集的ST37型菌株进行SNP分析,了解它们是否来源于同一克隆。同时将我们的菌株与公布了全基因组序列的ST37型菌株共同进行SNP分析,了解它们的来源。同时应用软件(https://cge.cbs.dtu.dk/services/ResFinder/)分析菌株基因组中的耐药基因。
1.2.5 菌株的MIC值测定
按CLSI标准采用琼脂稀释法测定抗菌药物对ST37型菌株的MIC值。
2 结果
2.1 一般资料
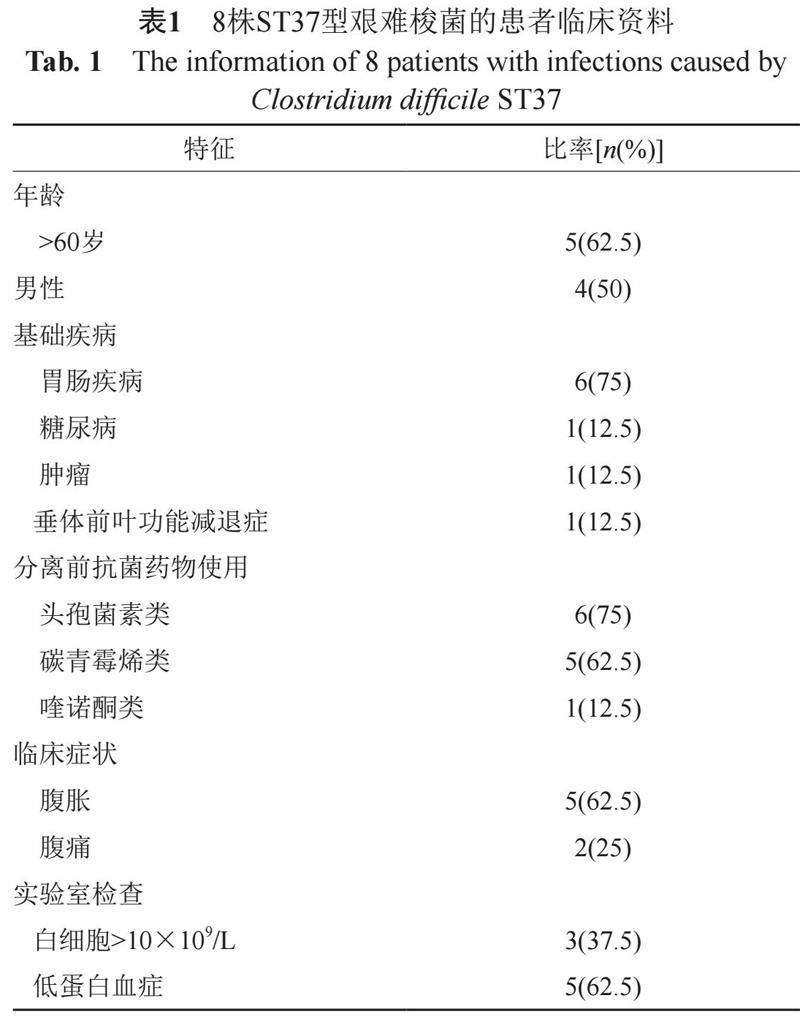
研究期间,124例腹泻患者粪便中共检测艰难梭菌31例,均为产毒性菌株, 其中tcdA+B+菌株23例(74.2%,23/31),经MLST分型可分为13个STs(ST2/3/8/35/54/208/209/210/211/212/213/214/218)。tcdA-B+菌株8例(25.8%,8/31),经MLST分型均为ST37型。这8株ST37菌株分别命名为(WCHCD109、WCHCD159、WCHCD770、WCHCD1216、WCHCD1262、WCHCD1450、WCHCD1577和WCHCD1641)。所有ST37菌株在分离前均使用过广谱抗生素,主要来源于老年患者,男女比例为1:1,临床腹泻症状较轻,伴有腹胀或腹痛。胃肠道疾病(包括乙肝肝硬化失代偿、重症胰腺炎、肠梗阻术后、肝移植术后)为主要的基础疾病(表1)。
2.2 全基因组测序结果
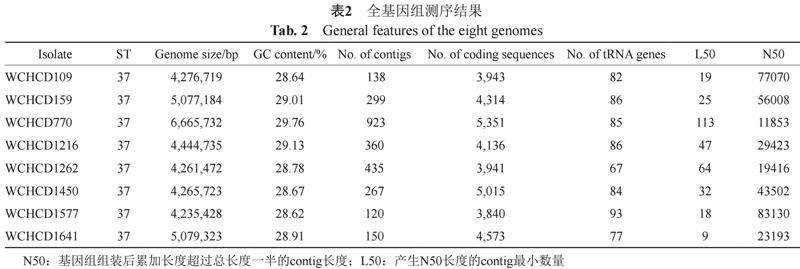
从表2结果中可以看出,这8株菌平均基因全长为5 Mb,平均GC含量为29%,最小的contig数目为120,平均基因预测4000个基因,且大部分在GenBank中能找到序列相同基因。
2.3 基因组SNP比较分析
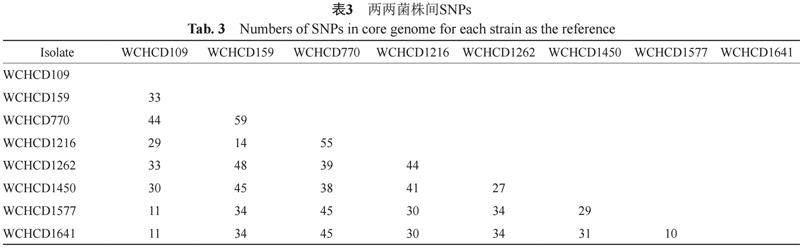
将这8株菌染色体基因中的核心基因组(约2Mb序列)除去在连接部位以及用Harvest软件没有标注“PASS”的,再进一步进行SNP分析,表3为两两菌株之间SNPs比较,根据临床资料,WCHCD770为最早收集的菌株,以其作为参照,图1代表其他菌株与WCHCD770之间的SNPs进化树。据艰难梭菌每年进化突变率,设定其SNP大于10时非近期传播事件。结果分析显示了菌株间的亲缘关系,可见WCHCD1577与WCHCD1641在同一分枝上,其SNPs为10,它们相互亲缘关系最近,可能来自同一克隆。而WCHCD770则与WCHCD14-159亲缘关系最远,SNPs为59。 由此可见,全基因组测序SNP分析能将同一MLST型菌株更准确地区分开来,也是克隆传播分型的最佳方法。
2.4 ST37型艰难梭菌来源分析
对已公布全基因组的艰难梭菌基因库659株菌進行MLST分析发现,共有8株为ST37,分别是:
DA00065 (ST37; clinical isolate, recovered in 2010, USA, GenBank accession no. AVIU00000000)
P71 (ST37; clinical isolate, recovered in 2009, USA, GenBank accession no. AVMW00000000);
P74 (ST37; clinical isolate, recovered in 2009, USA, GenBank accession no. AVMY00000000);
M68 (ST37; clinical isolate, recovered in 2006, Ireland, GenBank accession no. NC_017175);
E13 (ST37; clinical isolate, France, GenBank accession no. CAMF00000000);
002-P50-2011 (ST37; clinical isolate, USA, GenBank accession no. AGAA00000000);
050-P50-2011 (ST37; clinical isolate, USA GenBank accession no. AGAB00000000);
VL-0005 (ST37; clinical isolate, Canada, GenBank accession no. CZWV00000000)。
将基因库中的8株ST37 型菌与本研究收集的8株菌做致病性决定区(pathogenicity locus, PaLoc)比较,发现这些菌株的PaLoc完全相同,说明ST37型艰难梭菌的致病性决定基因相对保守。图2为这16株菌的SNP进化树分析。从图中可见WCHCD109、WCHCD159、WCHCD1216、WCHCD1577和WCHCD1641与基因库中2006年爱尔兰分离的M68亲缘关系最近,而WCHCD770、WCHCD1262和WCHCD1450与基因库中加拿大分离VL-0005亲缘关系最近。这也揭示了菌株的可能来源。
2.5 体外药敏结果
应用琼脂二倍稀释法测定抗菌药物对8株ST37型艰难梭菌的MIC值的分布范围,万古霉素(≤0.03~0.25 ?g/mL),克林霉素(8~>512 ?g/mL),利福平(≤0.03~>512 ?g/mL),莫西沙星(≤0.03~16 ?g/mL),甲硝唑(≤0.03 ?g/mL),四环素(≤0.03~32 ?g/mL) (表4)。
2.6 耐药基因分析结果
将本研究8株菌的全基因组序列在(https://cge.cbs.dtu.dk/services/ResFinder/)中分析其耐药基因(表5),8株艰难梭菌对克林霉素均耐药,有5株存在ermB耐药基因,但WCHCD109和WCHCD1577没有ermB耐药基因。5株艰难梭菌对四环素耐药,均存在tet耐药基因。4株艰难梭菌对利福平耐药,均存在rpoB耐药基因。4株艰难梭菌对莫西沙星耐药,存在gryA或gryB耐药基因。
3 讨论
CDI是目前公认的最重要的院内感染之一,近年来,其发病率病死率在全球呈上升趋势。艰难梭菌致病主要是由2个毒素基因tcdA和tcdB。欧美国家流行的艰难梭菌以tcdA+B+型菌株为主,亚洲以tcdA-B+型菌株为主。而ST37型为亚洲tcdA-B+型菌株主要的流行特征。本研究显示,tcdA-B+基因型的全部菌株为ST37型,这与既往ST37型在中国的分子流行病学报道一致[9-10]。药敏试验显示未发现万古霉素和甲硝唑耐药菌株,这与既往报道一致,提示CDAD治疗的一线用药仍是万古霉素和甲硝唑,但需要注意是目前艰难梭菌对这两种抗菌药物的敏感性呈逐年降低的趋势[11-12]。所有菌株均对克林霉素耐药,克林霉素耐药机制主要是由编码23SrRNA甲基化酶的erm基因所介导。但有2株(25%)未携带ermB基因,说明尚存在其他的耐药机制,是否存在外排泵、药物修饰酶、核糖体蛋白L4或L22的突变等,尚需要进一步研究。
分子分型是监测CDI感染暴發及病原溯源的一种非常重要的方法。目前艰难梭菌常用的分型方法包括脉冲场凝胶电泳(pulsed-field gel electrophoresis, PFGE)、PCR-核糖体分型、多位点序列分型(multilocus sequence typing, MLST)和多位点可变数目串联可重复序列分析(multiple-locus variable-number tandem- repeat analysis, MLVA)等[13]。但这些传统方法均存在一定局限性,如分辨力、重复性、可操作性和实验室间可比性等[13]。近年来,逐渐兴起的全基因组测序(whole genome sequenching, WGS)可准确研究细菌进化、流行途径和菌群分布情况,后续的单核苷酸多态性(single-nucleotide polymorphism,SNP)分析可区别菌株间单个核苷酸差异,因此具有更高的分辨力,有助于判别流行菌株是否来源于同一克隆[13]。就艰难梭菌而言,大量的研究表明,平均每个位点每年的突变率是1.47×10?7~5.33×10?7,相当于每个基因组每年的SNP大约为1~2[14-17]。因此,界定SNP值为0~2为近期克隆传播事件,SNP值大于10具有遗传差异的[14,18-19]。
本研究对8株同属ST37型tcdA-B+艰难梭菌进一步研究,探讨其是否存在院内传播,为进一步监测和预防医院感染爆发非常重要。研究通过WGS和SNP发现以最早分离的菌株WCHCD770作为参照,其他7株与之比较的SNP最小为38,可以认为它们与WCHCD770这株菌并非直接近期传播。结果表明相同ST型艰难梭菌感染,可能相互之间并没有进化关系,排除传播可能。而将这8株菌两两之间做SNP分析,发现WCHCD1577与WCHCD1641之间的SNP为10,虽然处于界定标准的边缘,结合相应临床资料,WCHCD1577分离菌患者出ICU的时间与WCHCD1641分离菌患者入ICU的时间仅相隔4 d,他们的住院床号相邻。而在院内感染的环节中,患者虽是传染源,但医务工作者处于这个医疗环境中,他们在接触患者时可通过手或医疗器械将芽孢定植而进行传播,易于成为潜在的传播者。通过采集医务人员的手部标本进行增菌培养,可推断该菌株在院内的传播是否是医务人员的手为媒介。推测这两株菌可能来源于同一克隆,患者所处的环境可能为其传播途径。因此,严格正确的手卫生、消毒和及时更换病房用品或医疗器械,有助于减少艰难梭菌的院内传播。将这8株菌的基因序列与基因库(Genbank)中已知的ST37型艰难梭菌基因组数据的SNP分析发现,WCHCD109、WCHCD159、WCHCD1216、WCHCD1577、WCHCD1641与爱尔兰克隆流行菌株M68非常接近,WCHCD770、WCHCD1262和WCHCD1450与加拿大菌株VL-0005也非常接近,推测它们之间可能存在远期传播事件。需要进一步了解这些患者的活动轨迹,以获取菌株来源更为准确的依据。
参 考 文 献
Benjamin H M, Horace R W. Clostridium difficile infection and antibiotic-associated diarrhoea[J]. Clin Med, 2018, 18(3): 237-241.
Leffler D A, Lamont J T. Clostridium difficile infection[J]. N Engl J Med, 2015, 372(16): 1539-1548.
Smits W K, Lyras D, Lacy D B, et al. Clostridium difficile infection[J]. Nat Rev Dis Primers, 2016, 2: 16020.
Cornelis W K, Elisabeth M T, Chris L, et al. Comparative analysis of an expanded Clostridium difficile reference strain collection reveals genetic diversity and evolution through six lineages[J]. Infect Genet Evol, 2012, 12(7): 1577-1585.
Hawkey P M, Marriott C, Liu W E, et al. Molecular epidemiology of Clostridium difficile infection in a major Chinese hospital: An underrecognized problem in Asia?[J]. J Clin Microbiol, 2013, 51(10): 3308-3313.
Drudy D, Fanning S, Kyne L. Toxin A-negative, toxin B-positive Clostridium difficile[J]. Int J Infect Dis, 2007, 11(1): 5-10.
Kim J, Pai H, Seo M R, Kang J O. Clinical and microbiologic characteristics of tcdA-negative variant Clostridium difficile infections[J]. BMC Infect Dis, 2012, 12: 109.
Haihui H, Shi W, Minggui W, et al. Clostridium difficile infections in a Shanghai hospital: Antimicrobial resistance, toxin profiles and ribotypes[J]. Int J Antimicrob Agents, 2009, 33(4): 339-342.
Chen Y B, Gu S L, Wei Z Q, et al. Molecular epidemiology of Clostridium difficile in a tertiary hospital of China[J]. J Med Microbiol, 2014, 63(4): 562-569.
Yan Q, Zhang J, Chen C, et al. Multilocus sequence typing (MLST) analysis of 104 Clostridium difficile strains isolated from China[J]. Epidemiol Infect, 2013, 141(1): 195-199.
Adler A, Miller-Roll T, Bradenstein R, et al. A national survey of the molecular epidemiology of Clostridium difficile in Israel: the dissemination of the ribotype 027 strain with reduced susceptibility to vancomycin and metronidazole[J]. Diagn Microbiol Infect Dis, 2015, 83(1): 21-24.
Kouhsari E, Douraghi M, Krutova M, et al. The emergence of metronidazole and vancomycin reduced susceptibility in Clostridium difficile isolates in Iran[J]. J Glob Antimicrob Resist, 2019, 18: 28-33.
李志荣, 赵建宏. 艰难梭菌分子分型方法研究进展[J]. 中国感染控制杂志, 2017, 16(4): 377-382.
Didelot X, Eyre D W, Cule M, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission[J]. Genome Biol, 2012, 13(12): R118.
Eyre D W, Cule M L, Wilson D J, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing[J]. N Eng J, Med, 2013, 369(13): 1195-1205.
He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile[J]. Nat. Genet, 2013, 45(1): 109-113.
Knetsch C W, Connor T R, Mutreja A, et al. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011[J]. Euro Surveill, 2014, 19(45): 20954.
Mac Aogáin M, Moloney G, Kilkenny S, et al. Whole-genome sequencing improves discrimination of relapse from reinfection and identifies transmission events among patients with recurrent Clostridium difficile infections[J]. J Hosp Infect, 2015, 90(2): 108-116.
Stone N E, Sidak-Loftis L. C, Sahl J W, et al. More than 50% of Clostridium difficile isolates from pet dogs in Flagstaff, USA, carry toxigenic genotypes[J]. PLos One, 2016, 11(10): e 0164504.
基金項目:四川省卫生健康科研课题(No. 19PJ223)
作者简介:陈熔,女,生于1981年,博士,主治医师,研究方向为感染性疾病的临床诊疗,E-mail: 38824605@qq.com
