高能固体推进剂粘合剂固化催化机理的密度泛函理论研究①
2023-04-26罗国勤桑丽鹏朱卫华
李 慧,罗国勤,桑丽鹏,朱卫华*
(1.南京理工大学 化学与化工学院,南京 210094;2.湖北航天化学技术研究所,襄阳 441003)
0 引言
复合固体推进剂是火箭、导弹等飞行器的推进系统中的重要能量源,其主要由无机氧化剂(如高氯酸铵、二硝酰胺铵等)、粘合剂、金属燃料、固化剂和其他添加剂等组分[1-3]。其中,具有一定粘性的高分子或者预聚物的粘合剂在体系起着关键作用,它可将多组分推进剂体系中的固体颗粒粘合聚结,赋予推进剂颗粒尺寸稳定性和结构完整性,并在燃烧过程中充当燃料[4-5]。例如,基于端羟基聚丁二烯(HTPB)、聚乙二醇(PEG)和缩水甘油叠氮聚醚(GAP)等的聚氨酯被广泛用作固体推进剂和聚合物粘结炸药中的预聚物粘合剂[6-7]。粘合剂中的端羟基基团与异氰酸酯基团反应生成氨基甲酸酯,进一步固化成三维网络结构的聚氨酯。其中,最常用有甲苯二异氰酸酯(TDI)[8]、异佛尔酮二异氰酸酯(IPDI)[9]和基于水改性的六次甲基-1,6-二异氰酸酯(又称缩二脲多异氰酸酯,N-100)[10-11]。由于这类反应的固化时间长且固化温度高,所以需要添加合适的催化剂提高其固化速度、降低固化温度以获得完全固化[11]。有机金属类化合物(如二月桂酸二丁基锡(DBTDL)[12]、三苯基铋(TPB)[13-14]和三-(乙氧基苯基)铋(TEPB)等)是适用于形成聚氨酯的催化剂。其中,关于三苯基铋的固化催化活性及其催化机理备受关注。谭惠民等[15]采用13C-NMR及二正丁胺滴定法研究了PEG与IPDI之间反应,提出了TPB催化下的反应机理。鲁国林等[14]发现TPB是IPDI-HTPB推进剂有效的固化催化剂,能明显降低固化温度和缩短固化时间。欧亚鹏等[16]采用非等温差示扫描量热法(DSC)探讨了催化剂TPB和TEPB对HTPB-TDI体系固化反应的影响。结果表明,TPB和TEPB均可降低固化反应的活化能,从而减小固化温差,缩短固化时间。虽然催化剂三苯基铋对推进剂粘合剂的固化催化机理已有了一些研究,但实验条件存在差异导致得到不同的结论,三苯基铋对端羟基类聚氨酯的催化机理尚未达到共识。然而,量子化学是揭示复杂反应机理的有效理论方法,具有从微观层次上提供化学反应基本信息的独特优势,对进一步详细了解反应机理有重要帮助。比如,基于粘合剂聚(3,3′-双叠氮甲基氧杂环丁烷)-四氢呋喃(PBT)与TDI固化反应的密度泛函理论(DFT)研究,揭示了不同反应路线的活化能,建立了反应动力学模型[17]。
本研究将运用DFT研究在催化剂TPB作用下固化基元反应机理,从微观层面上探讨TPB对固化基元反应机制的影响。具体地探讨了粘合剂PEG和GAP体系的固化反应机理,包括PEG/TDI、GAP/TDI、PEG/IPDI、GAP/IPDI、PEG/N-100和GAP/N-100体系,为进一步发展新型固体推进剂粘合剂提供理论支持。
1 计算方法
本工作中的部分计算使用Gaussian 09程序[18]。采用(U)DFT-M06-2X/def2-SVP[19]理论方法对气相中的分子进行结构优化和频率分析。自由基结构的计算则采用非限制性开壳层方法(U)进行。通过频率分析验证每个优化结构是过渡态还是极小点,过渡态结构只有一个虚频的存在,并运用内禀反应坐标(intrinsic reaction coordinate, IRC)方法确认从每个过渡态连接到相应反应物或产物。
运用Materials Studio 7.0 软件中DMol3模块研究催化剂对体系固化反应的影响。首先,采用B3LYP和DFT-D色散校正方法对反应物和生成物的几何结构进行优化。然后基于优化的结构,通过将线性同步转变(LST)和二次同步转变(QST)方法相结合的LST/QST算法来筛选在无或有催化剂TPB参与下各种固化基元反应的过渡态,并得到相应的反应活化势垒。采用周期性体系进行过渡态搜寻,相应的晶格参数为50 Å×50 Å×50 Å。
2 结果与讨论
2.1 结构选取
2.1.1 PEG和GAP链节长度的选择
固化反应是指粘合剂的端羟基(—OH)与固化剂的异氰酸酯(—NCO)亲核加成形成了氨基甲酸酯基,如图1所示。在这个反应过程中,影响反应的速率和难易程度的一步是聚醇类端羟基的断裂。
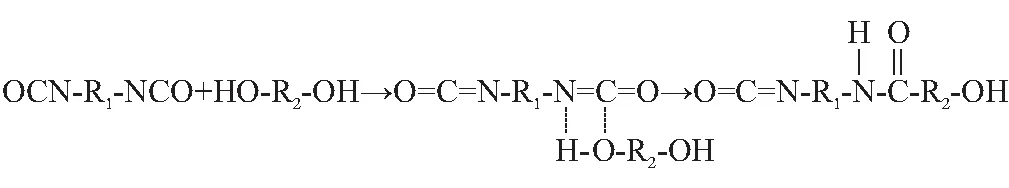
图1 固化基元反应示意图
考虑到所选取聚醇类的结构单元的聚合度n对固化反应能垒的影响,这里计算了具有不同聚合度(n=1、2、3、4和5)的PEG和GAP中参与反应的O—H键的键解离能。键离解能(Bond dissociation energy,D)一般被定义为分子中化学键断裂过程的反应焓变。它反映了键断裂过程所需要的能量,其数值越大,则分子越稳定。其计算公式由式(1)给出:
D(A-B)=H(A)+H(B)-H(AB)
(1)
式中H(A)和H(B)分别为分子AB生成两个自由基片段A和B的焓值;H(AB)为分子AB的焓值。
在(U)M06-2X/def2-SVP水平上,对研究的分子或者自由基进行结构优化和频率分析。通过计算热化学性质得到反应焓变,即键解离能D如表1所示。随着聚合度n增加,PEG和GAP的键离解能数值变化不大,PEG和GAP链节长短对固化反应的影响较小。为平衡计算效率,选择了两个结构单元链节作为PEG和GAP的研究模型。

表1 PEG和GAP中参与反应的O—H键的键离解能
2.1.2 TDI异构体的选择
固化剂TDI主要有两种异构体:2,4-TDI和2,6-TDI,均含有异氰酸酯基团—NCO。所以,TDI反应时存在不同的反应位点。基于M06-2X/def2-SVP方法,计算了不同反应位点下PEG/TDI和GAP/TDI反应路径的过渡态。图2展示了两种TDI异构体分别与GAP和PEG反应的过渡态。

图2 不同反应位点下GAP/TDI和PEG/TDI反应路径的过渡态(过渡态能量以反应物能量为相对值;键长单位:Å)
由图2可以看出,GAP伯醇上带负电的O原子直接进攻2,4-TDI中—NCO上带正电的C原子,同时伯醇的H原子逐渐靠近—NCO上N原子,形成一个四元环过渡态。其次,异构体2,4-TDI的反应能垒ΔE比异构体2,6-TDI更低,更容易发生反应。例如,PEG的端羟基与2,4-TDI对位—NCO的反应能垒ΔE为24.69 kcal/mol。因此,在讨论有无催化剂条件下,主要考虑2,4-TDI的基元反应。
2.2 PEG/IPDI/TPB与GAP/IPDI/TPB
采用DMol3程序中B3LYP方法,计算了PEG/IPDI与PEG/IPDI/TPB的反应路径势能面和相应过渡态结构(图3)。

(a)Potential energy surfaces of the reaction paths of PEG/IPDI and PEG/IPDI/TPB
首先,PEG上端羟基O—H键的键长变长。然后,H和O会分别进攻IPDI中—NCO上的N和C原子,进而形成四元环状过渡态(N—C—O—H),其中N—H和C—O键键长分别为1.452 Å和2.195 Å,最后生成氨基甲酸酯。加入催化剂TPB后,由于PEG会靠近TPB分子,四元环状过渡态中N—H键键长被拉长至1.907 Å,铋原子与羟基氧络合后使羟基质子活化。活化的羟基质子会加快与异氰酸酯—NCO络合的速率,促进产物氨基甲酸酯的形成。这一结论和实验提出的猜想吻合[16]。由图3中反应路径的势能面可见,在没有催化剂TPB时,基元反应PEG/IPDI的反应能垒为46.58 kcal/mol,反应放热为17.68 kcal/mol。加入催化剂TPB后,反应能垒降低至31.32 kcal/mol,反应放热为18.79 kcal/mol。因此,加入催化剂TPB后降低了能垒,有利于提高反应活性。
GAP/IPDI/TPB反应的过渡态和能量势垒如图4所示。同样,在催化剂TPB的作用下,铋原子会靠近GAP端羟基的O—H键,四元环状过渡态的N—H键的键长从1.176 Å(图4(a))拉长至1.645 Å(图4(b))。随后,铋原子与O—H键的质子氢发生络合作用,从而导致O—H键的键长逐渐增加,H原子更容易向异氰酸酯基团上的N原子靠近,而分离的O原子则进攻异氰酸酯基团上的C原子,逐渐形成氨基甲酸酯结构,最终交联为聚氨酯产物。这是一个放热过程,其中反应放热为11.78 kcal/mol,反应能垒为20.62 kcal/mol。此外,通过对比催化剂TPB分别参与PEG/IPDI和GAP/IPDI前后的反应能垒的下降幅度,发现GAP/IPDI的反应能垒降低得更明显,反应放热也增加了6.89 kcal/mol。
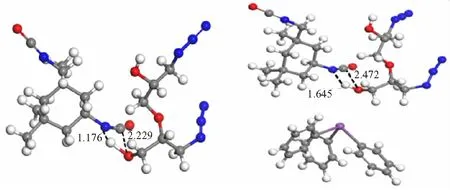
(a)The transition state of GAP/IPDI (b)The transition state of GAP/IPDI/TPB
2.3 PEG/N-100/TPB 与GAP/N-100/TPB
选取二元醇PEG和GAP分别与多异氰酸酯N-100反应,通过搜索PEG/N-100/TPB反应与GAP/N-100/TPB反应的过渡态,确定了两种基元反应的反应路径势能面(见图5)。
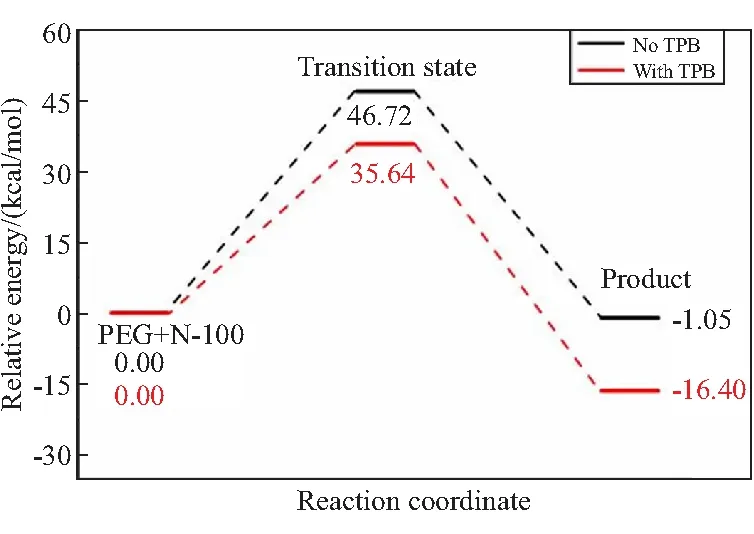
(a)PEG/N-100/TPB
GAP/N-100的反应能垒为46.84 kcal/mol,略微大于PEG/N-100(46.72 kcal/mol)。在催化剂TPB参与下PEG/N-100/和GAP/N-100的反应能垒均有明显降低。其中,GAP/N-100的反应能垒从46.84 kcal/mol降低至35.95 kcal/mol,反应放热增多了7.26 kcal/mol。其次,反应路径的过渡态结构中主要的键长参数如图5所示。GAP/N-100/TPB反应的过渡态中O—H、N—H和C—O键的距离分别为1.022、1.956、2.624 Å,与PEG/N-100/TPB的过渡态相比,O—H和C—O键都分别被拉长,这些键长的变化有可能会导致活化能垒的增加。
2.4 PEG/2,4-TDI/TPB与GAP/2,4-TDI/TPB
图6是PEG/2,4-TDI/TPB和GAP/2,4-TDI/TPB的反应路径能量图。
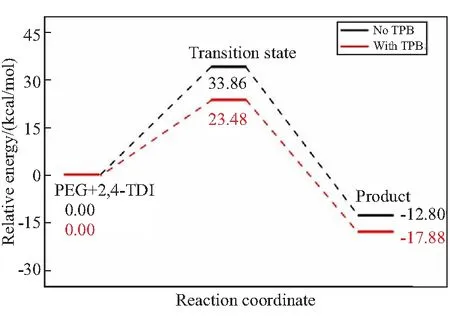
(a)PEG/2,4-TDI/TPB
由图6可以看出,加入催化剂TPB后,PEG/2,4-TDI/TPB和GAP/2,4-TDI/TPB两个固化基元反应的能垒显著地降低。在没有催化剂TPB时,基元反应PEG/2,4-TDI的能垒为33.86 kcal/mol,反应放热为12.80 kcal/mol。加入催化剂TPB后,反应能垒为 23.48 kcal/mol,反应放热为17.88 kcal/mol。此外,通过对比PEG/2,4-TDI/TPB和GAP/2,4-TDI/TPB反应活化能垒的下降幅度,发现GAP/2,4-TDI的反应能垒降低了12.80 kcal/mol,而反应放热增加了19.39 kcal/mol。因此,催化剂TPB的参与对粘合剂GAP体系影响更大。
3 结论
本论文采用DFT方法,研究了复合固体推进剂中粘合剂GAP和PEG分别与固化剂(TDI、IPDI和N100)之间的基元反应机理,并且讨论催化剂三苯基铋对固化基元反应的影响。主要结论如下:
(1)基于粘合剂GAP和PEG键离解能的计算,发现链节长短对固化基元反应的影响不大。因此,采用两个结构单元的GAP和PEG进行研究。
(2)催化剂三苯基铋的参与明显地降低了基元反应的活化能垒,增加反应放热。催化剂TPB的加入,对固化基元反应具有显著的影响。
(3)三苯基铋对固化基元反应的催化机制:在催化剂TPB的作用下,铋原子会靠近端羟基O—H键。铋原子与O—H键的质子氢发生络合作用,使羟基质子活化,加快H原子向异氰酸酯基团上的N原子移动,而分离的O原子迅速进攻异氰酸酯基团上的C原子并结合,从而提高氨基甲酸酯结构的形成速率。
